변분 양자 고유값 분해기 (VQE)
이 모듈을 진행하려면 Python 환경이 갖춰져 있어야 하며, 아래 패키지들의 최신 버전이 설치되어 있어야 합니다:
qiskitqiskit_ibm_runtimeqiskit-aerqiskit.visualizationnumpypylatexenc
패키지 설치 및 환경 설정은 Qiskit 설치 가이드를 참고하세요. 실제 양자 컴퓨터에서 작업을 실행하려면 IBM Cloud 계정 설정 가이드의 단계에 따라 IBM Cloud 계정을 만들어야 합니다.
이 모듈은 테스트되었으며, 약 8분의 QPU 시간을 사용했습니다. 이는 추정치이며, 실제 사용량은 다를 수 있습니다.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-aer qiskit-ibm-runtime scipy
# Uncomment and modify this line as needed to install dependencies
#!pip install 'qiskit>=2.1.0' 'qiskit-ibm-runtime>=0.40.1' 'qiskit-aer>=0.17.0' 'numpy' 'pylatexenc'
소개
20세기 초 양자역학 모델이 개발된 이후, 과학자들은 전자가 원자핵 주위를 고정된 궤도로 돌지 않고, 오비탈(orbital)이라 불리는 확률 영역에 존재한다는 것을 알게 되었습니다. 이 오비탈들은 전자가 점유할 수 있는 특정하고 불연속적인 에너지 준위에 해당합니다. 전자는 자연적으로 바닥 상태(ground state)라고 하는 가장 낮은 에너지 준위에 있으려 합니다. 그러나 전자가 충분한 에너지를 흡수하면 더 높은 에너지 준위로 도약하여 들뜬 상태(excited state)에 진입할 수 있습니다. 이 들뜬 상태는 일시적이며, 전자는 결국 더 낮은 에너지 준위로 돌아오면서 흡수했던 에너지를 주로 빛의 형태로 방출합니다. 이 에너지 흡수와 방출의 기본 과정은 원자들이 어떻게 상호작용하고 결합을 형성하는지 이해하는 데 중요합니다.
원자들이 모여 분자를 형성할 때, 원자 오비탈들이 결합하여 분자 오비탈을 형성합니다. 이 분자 오비탈 내 전자들의 배열과 에너지 준위는 생성된 분자의 특성과 화학 결합의 강도를 결정합니다. 예를 들어, 두 개의 수소 원자로부터 수소 분자()가 형성될 때, 각 원자의 전자는 원자 오비탈을 점유합니다. 원자들이 서로 가까워지면 이 원자 오비탈들이 겹치고 결합하여 새로운 분자 오비탈을 형성합니다 — 하나는 에너지가 낮은 결합 오비탈(bonding orbital), 다른 하나는 에너지가 높은 반결합 오비탈(anti-bonding orbital)입니다. 각 수소 원자에서 온 두 전자는 에너지가 낮은 결합 오비탈을 우선적으로 점유하게 되어 분자를 묶어주는 안정한 공유 결합이 형성됩니다. 분리된 원자들과 형성된 분자 사이의 에너지 차이, 특히 분자 오비탈 내 전자들의 에너지는 결합의 안정성과 특성을 결정합니다.
이어지는 절에서는 분자에 초점을 맞추어 이 분자 형성 과정을 살펴볼 것입니다. 실제 양자 컴퓨터와 고전 최적화 기법을 함께 사용하여, 이 단순하지만 근본적인 과정의 에너지를 구할 것입니다. 이 실험은 양자 계산이 계산 화학 문제 해결에 어떻게 응용될 수 있는지, 그리고 전자 에너지의 역할에 대한 통찰을 실제로 보여줍니다.
VQE - 고유값 문제를 위한 변분 양자 알고리즘
화학을 위한 근사 기법 - 변분 원리와 기저 집합
에르빈 슈뢰딩거(Erwin Schrödinger)의 양자역학에 대한 기여는 새로운 전자 모델을 도입한 것에 그치지 않습니다. 그는 유명한 시간 의존 슈뢰딩거 방정식을 개발함으로써 파동역학의 기초를 확립했습니다:
여기서 는 계의 전체 에너지를 나타내는 해밀토니안 연산자이고, 는 계의 양자 상태에 대한 모든 정보를 담고 있는 파동 함수입니다. (참고: 는 전체 시간 미분이며, 에너지 고유값 는 여기에 명시적으로 포함하지 않습니다.)
그러나 원자와 분자의 허용 에너지 준위를 구하는 것과 같은 많은 실용적인 응용에서는 대신 시간 독립 슈뢰딩거 방정식(에너지 고유값 방정식)을 사용합니다. 이는 시간 의존 형태에서 정상 상태(stationary state)를 가정함으로써 유도됩니다. 정상 상태란 공간의 특정 지점에서 입자를 발견할 확률 밀도가 시간에 따라 변하지 않는 양자 상태입니다.
이 형태에서 는 양자 상태 에 대응하는 에너지 고유값을 나타냅니다. 해밀토니안에는 전자와 원자핵의 운동 에너지, 전자와 원자핵 사이의 인력, 전자들 사이의 척력 등 다양한 에너지 기여가 포함됩니다.
에너지 고유값 방정식을 풀면 원자 및 분자 계의 양자화된 에너지 준위를 계산할 수 있습니다. 그러나 분자의 경우, 전자의 공간 분포를 기술하는 파동 함수 가 복잡하고 고차원이기 때문에 이를 정확하게 풀기가 어렵습니다.
따라서 과학자들은 실용적이고 정확한 해를 얻기 위해 근사 기법을 사용합니다. 이 연구에서는 두 가지 핵심 방법에 초점을 맞춥니다:
-
변분 원리
이 방법은 파동 함수를 근사하고, 목표 에너지(보통 계의 바닥 상태 에너지)에 최대한 가깝게 조정합니다. 변분 원리의 핵심 아이디어는 간단합니다:
- 파동 함수 ("시도 함수")을 추측하면, 이로부터 계산된 에너지는 항상 계의 바닥 상태 에너지()와 같거나 더 높습니다.
- 시도 함수의 매개변수 , 즉 를 조정함으로써 바닥 상태 에너지에 점점 더 가까운 근사값을 얻을 수 있습니다.
- 정확도는 시도 파동 함수 의 선택에 크게 의존합니다. 잘못 선택된 시도 함수는 실제 값과 동떨어진 에너지 추정값을 낼 수 있습니다.
-
기저 집합 근사
두 번째 근사 방법은 파동 함수를 구성하는 단계인 기저 집합(basis set) 접근법입니다. 양자 화학에서 분자의 슈뢰딩거 방정식을 정확하게 푸는 것은 거의 불가능합니다. 대신, 복잡한 다전자 파동 함수를 더 단순하고 미리 정의된 수학 함수들로 구축하여 근사합니다. 기저 집합이란 분자 내 원자들을 중심으로 배치된 이러한 알려진 수학 함수들의 모음으로, 계 내 전자들의 형태와 거동을 표현하기 위한 구성 요소로 사용됩니다. 마치 표준 레고 블록만으로 정교한 조각품을 재현하려는 것과 같습니다 — 블록의 종류와 크기가 많을수록(기저 집합이 클수록) 원래 형태를 더 정확하게 근사할 수 있습니다.
이 기저 함수들은 종종 수소 원자와 같은 단순한 계의 해석적 해에서 영감을 받아, 가우스형(Gaussian) 또는 슬레이터형(Slater-type) 함수의 형태를 취합니다. 이론적으로 "정확"하지만 다루기 어려운 완전한 분자 오비탈을 직접 다루는 대신, 이 기저 함수들의 선형 결합(계수가 있는 합)으로 표현합니다. 기저 함수가 원자 오비탈과 유사한 경우 이 방법을 원자 오비탈의 선형 결합(LCAO, Linear Combination of Atomic Orbitals) 접근법이라고 합니다. 이 선형 결합의 계수를 최적화함으로써, 선택한 기저 집합의 제약 내에서 가능한 최선의 근사 파동 함수와 에너지를 찾을 수 있습니다.
- 기저 집합에 포함된 함수가 많을수록 근사가 더 좋아지지만, 그만큼 더 높은 계산 비용이 필요합니다.
- 작은 기저 집합은 대략적인 추정값을 제공하는 반면, 큰 기저 집합은 더 많은 계산 자원이 필요하지만 더 정밀한 결과를 줍니다.
요약하면, 계산을 실행 가능하게 하고 계산 비용을 줄이기 위해 변분 원리를 사용하여 파동 함수를 근사하며, 이를 통해 계산 복잡성을 줄이고 에너지를 최소화하는 반복 최적화가 가능합니다. 한편, 기저 집합 접근법은 연속적인 파동 함수를 직접 풀지 않고 원자 오비탈을 미리 정의된 함수들의 조합으로 표현함으로써 계산을 단순화합니다.
이해도 확인
시도 파동 함수 를 고려하세요. 여기서 는 규격화 상수이고 는 조정 가능한 매개변수입니다.
(a) 다음 조건을 만족하는 를 구하여 시도 파동 함수를 규격화하세요. .
Answer
주어진 시도 파동 함수를 규격화하면:
가우스 적분을 이용합니다:
로 놓으면:
(b) 다음과 같이 주어진 해밀토니안 의 기댓값을 계산하세요. 여기서 로, 단순 조화 진동자 퍼텐셜에 해당합니다.
Answer
조화 진동자의 해밀토니안은 다음과 같습니다:
운동 에너지 기댓값
2차 도함수를 구하면:
따라서:
표준 가우스 적분 결과를 이용하면:
퍼텐셜 에너지 기댓값
다음 공식을 이용합니다:
그러면:
전체 에너지 기댓값
(c) 를 최소화하여 변분 원리로 최적 를 구하세요.
Answer
에너지 최소화를 위해 를 최적화합니다.
미분
풀면:
를 에 대입하면:
이는 정확한 양자 조화 진동자 바닥 상태 에너지와 일치합니다.
VQE (변분 양자 고유값 분해기)
변분 양자 고유값 분해기(VQE)는 반응을 탐구하는 데 사용할 주요 방법입니다. 여기서는 VQE가 무엇인지, 어떻게 작동하는지 살펴보겠습니다. 그 전에 먼저 아래의 확인 문제를 통해 중요한 사항을 짚어보겠습니다.
이해도 확인
이미 화학 문제를 위한 전략이 많이 있는데, 왜 양자 컴퓨터가 필요한가요? 그리고 양자 컴퓨터와 고전 컴퓨터를 함께 사용하는 목적은 무엇인가요?
Answer
양자 컴퓨팅은 양자 상태의 지수적 증가로 인해 고전 컴퓨터가 어려움을 겪는 문제들을 해결함으로써 화학 분야에 혁신을 가져올 가능성이 있습니다. 리처드 파인만(Richard Feynman)은 자연을 시뮬레이션하려면 계산 자체도 양자적이어야 한다고 유명하게 언급한 바 있습니다 [참고 1].
예를 들어, 가장 단순한 기저 집합(STO-3G)으로 카페인을 시뮬레이션하려면 비트가 필요하며, 이는 관측 가능한 우주의 별 개수()보다 훨씬 큰 값입니다 [참고 2]. 반면 양자 컴퓨터는 160개의 Qubit으로 카페인의 전자 오비탈을 표현할 수 있습니다.
양자 컴퓨터는 중첩(superposition)과 얽힘(entanglement)을 이용해 양자 상호작용을 자연스럽게 처리하므로, 정확한 분자 시뮬레이션을 가능하게 하는 유망한 방법을 제공합니다. 또한 양자 컴퓨터(전자 시뮬레이션)와 고전 컴퓨터(데이터 전처리/후처리, 알고리즘 프로세스 관리, 최적화 등)의 장점을 결합할 수 있습니다. 이를 통해 재료 발견, 신약 설계, 반응 예측을 향상시키고 비용이 많이 드는 시행착오 실험을 줄일 것으로 기대됩니다. [참고 3][참고 4]
양자 컴퓨터가 화학 문제에 필요한 이유와 양자·고전 컴퓨팅 자원을 함께 사용하는 이유에 대해 더 알고 싶다면 다음 논문들을 참고하세요:
이제 VQE로 돌아가겠습니다.
VQE는 양자 컴퓨터와 고전 컴퓨터의 강점을 결합하며, 근본적으로 변분 원리를 활용하여 시스템의 바닥 상태 에너지를 구합니다. VQE를 이해하기 위해 세 가지 부분으로 나누어 살펴보겠습니다:
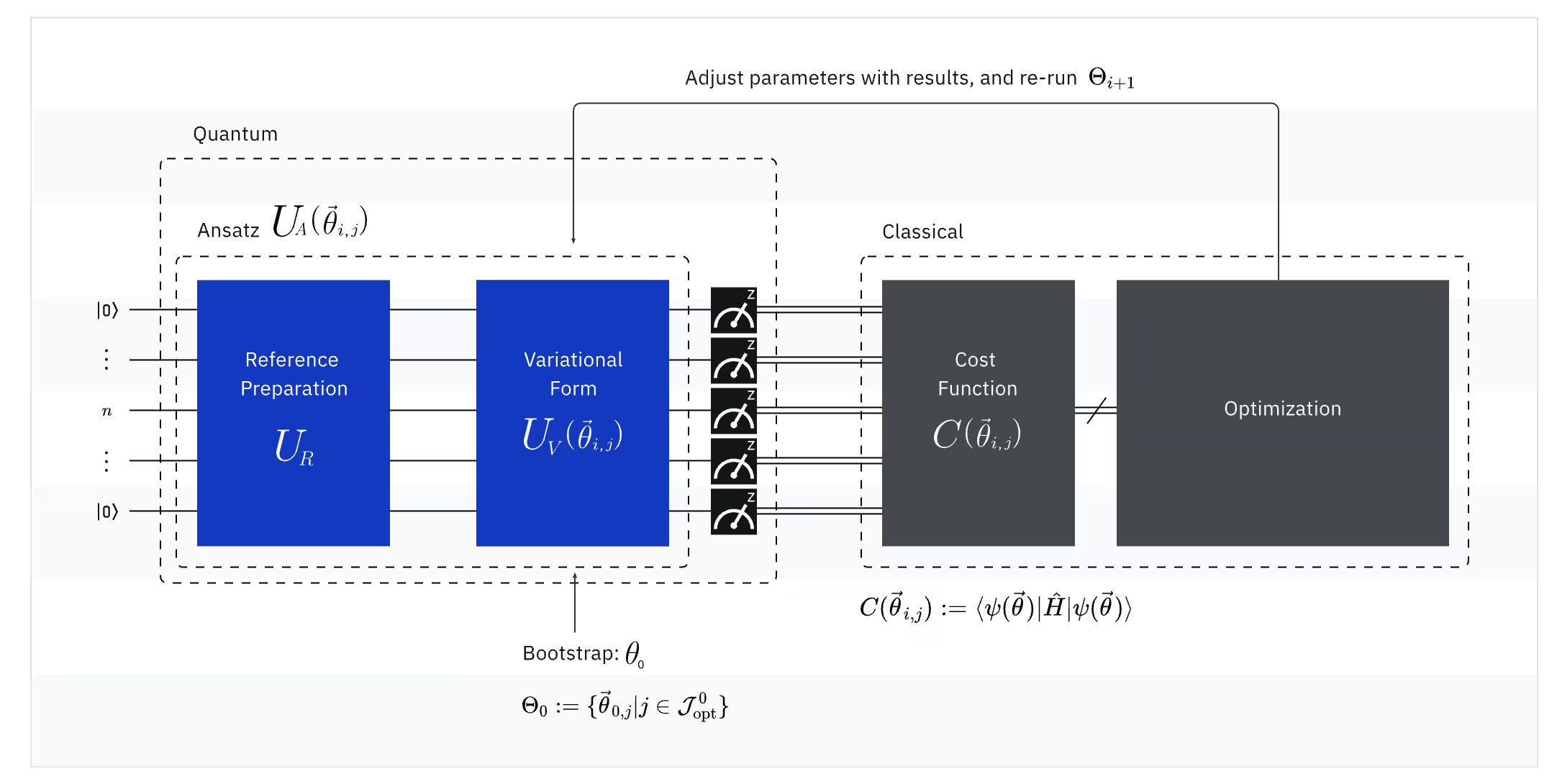
(양자) 관측량: 분자 해밀토니안 (분자의 에너지)
VQE에서 분자/원자 해밀토니안은 관측량(observable)으로, 실험을 통해 그 값을 측정할 수 있습니다. 우리의 목표는 분자의 가능한 최저 에너지(바닥 상태 에너지)를 찾는 것입니다. 이를 위해 매개변수화된 양자 Circuit(ansatz)으로 생성된 시험 양자 상태를 사용합니다. 관측량을 측정하고 가능한 최저 에너지에 도달할 때까지 양자 상태를 최적화합니다.
분자 해밀토니안에 사용되는 기저 집합은 필요한 qubit 수를 결정하며, VQE의 정확도에 직접적인 영향을 미칩니다. 올바른 기저 집합을 선택하는 것은 효율성과 정밀도의 균형을 맞추는 데 매우 중요합니다. 기저 집합을 변경하지 않고 계산을 단순화하기 위해, 대칭성 부과(symmetry imposition)와 활성 공간 축소(active space reduction) 같은 전략을 사용할 수 있습니다. 많은 분자는 대칭적인 형태(나비나 눈송이처럼)를 가지므로 일부 부분이 동일하게 작동합니다. 모든 부분을 개별적으로 계산하는 대신 고유한 부분에만 집중하면 양자 자원을 절약할 수 있으며, 이것이 대칭성 활용의 핵심입니다. 활성 공간 축소에서는 중요한 오비탈만 고려합니다. 모든 전자가 분자 에너지에 크게 영향을 미치는 것은 아니기 때문입니다. 핵에 가까운 전자는 대부분 변하지 않는 반면, 다른 전자들은 결합에 영향을 줍니다. 이러한 방법들을 적용하면 정확도를 유지하면서 VQE를 더 효율적으로 만들 수 있습니다.
적절한 기저 집합과 위의 전략들을 사용하여 분자 해밀토니안을 얻은 후에는, 이 해밀토니안을 양자 컴퓨터에 적합한 형태로 변환해야 합니다. 문제를 파울리(Pauli) 연산자에 매핑하는 것은 꽤 복잡할 수 있습니다. 특히 양자 화학에서는 구별 불가능한 입자(전자)를 다루는 반면, Qubit은 구별 가능하기 때문입니다. 여기서는 매핑의 세부 사항을 다루지 않겠지만, 다음 자료들을 참고해 주세요. 문제를 양자 연산자에 매핑하는 방법에 대한 일반적인 설명은 실전 양자 컴퓨팅에서 찾을 수 있습니다. 화학 문제를 양자 연산자에 매핑하는 방법에 대한 더 자세한 내용은 VQE를 활용한 양자 화학에서 확인할 수 있습니다.
이 모듈에서는 양자 컴퓨터 사용에 집중할 수 있도록 와 에 대한 적절한 (단일 qubit) 해밀토니안을 제공합니다. 이 단일 qubit 해밀토니안들은 STO-6G 기저 집합과 조던-위그너(Jordan-Wigner) 매핑을 사용하여 준비되었습니다. 조던-위그너 매핑은 하나의 스핀 오비탈의 점유를 하나의 Qubit의 점유에 대응시키기 때문에, 가장 간단한 물리적 해석을 갖는 가장 직관적인 매핑입니다. 또한 해밀토니안의 대칭성을 이용한 qubit 축소 기법을 사용하였는데, 이는 스핀 점유가 어떻게 행동하는지의 패턴을 활용하여 qubit 수를 줄입니다. 분자의 경우, 두 수소 원자 사이의 거리는 0.735 로 가정합니다.
(양자) Ansatz: 시험 파동 함수 (양자 Circuit으로 시험 양자 상태를 구성하는 방법)
VQE에서 ansatz(복수형: ansätze)는 두 가지 핵심 요소로 구성됩니다. 첫 번째는 초기 상태 준비로, 변분 매개변수 없이 양자 Gate를 적용하여 Qubit의 상태를 설정합니다. 두 번째 요소는 매개변수화된 양자 Circuit으로, 라디오의 다이얼처럼 조절 가능한 매개변수를 가진 특수한 양자 Circuit입니다. 이 매개변수들은 마지막 부분인 고전 최적화기(classical optimizer)에서 최적의 바닥 상태에 도달하기 위해 사용됩니다.
변분 원리 절에서 시험 상태의 품질이 변분 알고리즘 결과의 품질에 영향을 미친다는 것을 배웠습니다. 즉, 좋은 ansatz를 선택하는 것이 VQE에서 중요합니다. 이 또한 풍부하고 복잡한 주제입니다. 여기서는 다양한 유형의 ansatz나 그 기원에 대해 다루지 않겠습니다. 매개변수화된 양자 Circuit과 ansatz에 대해 더 알고 싶다면, 다양한 ansätze에 대한 자세한 설명과 예시를 제공하는 변분 알고리즘 설계 과정의 Ansatz 및 변분 형식 수업을 참고하세요.
이 모듈에서는 단일 qubit 해밀토니안을 사용할 것이므로, ansatz로 단일 qubit 매개변수화 양자 Circuit이 필요합니다. 다음 절에서는 세 가지 유형의 단일 qubit ansätze를 살펴봅니다. 이들을 비교하고 ansatz 선택 시 핵심 고려 사항을 논의하겠습니다.
(고전) Optimizer: 양자 Circuit 미세 조정
양자 컴퓨터가 ansatz로부터 관측량의 에너지를 측정하면, ansatz의 매개변수와 에너지 값이 고전 최적화기(classical optimizer)로 전송되어 조정됩니다. 이 최적화 과정은 일반적으로 SciPy와 같은 범용 과학 패키지를 사용하여 고전 컴퓨터에서 수행됩니다.
고전 최적화기는 측정된 에너지를 비용 함수(cost function)로 취급합니다. 최적화 문제에서 비용 함수(목적 함수라고도 함)는 특정 해결책이 얼마나 "좋은지"를 측정하는 수학 함수입니다. 최적화기의 목표는 이 비용 함수를 최소화하는 매개변수 집합을 찾는 것입니다. 분자의 바닥 상태 에너지를 찾는 맥락에서, 에너지 자체가 비용 함수 역할을 합니다 — 가능한 최저 에너지를 산출하는 양자 Circuit(우리의 "해결책")의 매개변수를 찾고자 합니다. 고전 최적화기는 이 측정된 에너지 값(비용)을 사용하여 양자 ansatz의 다음 최적화 매개변수 집합을 결정합니다. 업데이트된 매개변수들은 다시 양자 Circuit으로 전송되고 과정이 반복됩니다. 각 반복마다 고전 최적화기는 매개변수를 조정하여 에너지를 낮추려(비용 함수를 최소화) 시도하며, 사전 정의된 수렴 기준이 충족될 때까지 계속됩니다. 이상적으로는 가능한 최저 에너지(해당 결합 거리와 기저 집합에서 분자의 바닥 상태에 해당)를 찾게 됩니다.
SciPy와 같은 과학 패키지들은 다양한 최적화 전략을 제공합니다. 변분 알고리즘 설계 과정의 최적화 루프 수업에서 더 많은 내용을 찾아볼 수 있습니다. 여기서는 복잡한 에너지 지형에 적합한 최적화 알고리즘인 COBYLA(선형 근사에 의한 제약 최적화, Constrained Optimization BY Linear Approximations)를 사용합니다. 특히 COBYLA는 연구 대상 함수의 기울기를 계산하려 하지 않는데, 이를 기울기 없는 최적화기(gradient-free optimizer)라고 합니다. 눈을 감고 산악 지형에서 가장 높은 봉우리를 찾으려 한다고 상상해보세요. 전체 지형을 볼 수 없으므로 여러 방향으로 작은 걸음을 내딛으며 올라가고 있는지 내려가고 있는지 확인합니다. COBYLA는 이와 비슷한 방식으로 작동합니다 — 매개변수 공간을 이동하며 다양한 값을 테스트하고, 최선의 결과를 찾을 때까지 점진적으로 결과를 개선합니다.
이제 VQE 계산을 수행할 준비가 되었습니다. 이를 위해 아래의 확인 문제를 풀어보세요. 이 문제는 전체 과정을 요약합니다.
이해도 확인
빈칸에 알맞은 용어를 채워 VQE 과정 요약을 완성하고, 클릭하여 정답을 확인하세요.
VQE는 변분 양자 알고리즘으로, (1) ________ 과 고전 컴퓨팅의 강점을 결합하여 분자의 (2) __________ 를 찾는 데 사용됩니다. 과정은 시스템의 총 에너지를 나타내며 양자 측정에서 관측량 역할을 하는 (3) __________ 을 정의하는 것으로 시작됩니다. 다음으로, 분자의 시험 파동 함수를 나타내는 조절 가능한 매개변수가 있는 양자 Circuit인 (4) __________ 를 준비합니다. 이 매개변수들은 측정된 에너지를 최소화하기 위해 반복적으로 매개변수를 조정하는 고전 알고리즘인 (5) __________ 를 사용하여 최적화됩니다. 위의 논의에서는 미분 계산 없이 ansatz 매개변수를 개선하는 (6) __________ 최적화기를 사용했습니다. 분자의 가능한 최저 에너지를 찾았음을 의미하는 (7) __________ 에 도달할 때까지 과정이 계속됩니다.
단어 목록:
- classical optimizer
- ground state energy
- hardware-efficient
- ansatz
- molecular Hamiltonian
- COBYLA
- quantum computing
- convergence
Answer
1 → quantum computing
2 → ground state energy
3 → molecular Hamiltonian
4 → ansatz
5 → classical optimizer
6 → COBYLA
7 → convergence
VQE로 수소 원자의 바닥 상태 에너지 계산하기
이제 지금까지 배운 내용을 활용하여 수소 원자의 바닥 상태 에너지를 계산해 보겠습니다. 이 모듈 전반에 걸쳐 "Qiskit 패턴(Qiskit patterns)"이라는 양자 컴퓨팅 프레임워크를 사용합니다. 이 프레임워크는 워크플로를 다음과 같은 단계로 구분합니다:
- 1단계: 고전적 입력을 양자 문제로 매핑
- 2단계: 양자 실행을 위한 문제 최적화
- 3단계: Qiskit Runtime 기본 요소(Primitive)를 사용하여 실행
- 4단계: 후처리 및 고전적 분석

일반적으로 이 단계를 따라 진행할 것입니다.
먼저 Qiskit Runtime 기본 요소를 포함한 필요한 패키지들을 불러오겠습니다. 또한 현재 사용 가능한 양자 컴퓨터 중 가장 여유 있는 것을 선택합니다.
아래에는 처음 사용 시 자격 증명을 저장하는 코드가 포함되어 있습니다. 자격 증명을 환경에 저장한 후에는 반드시 노트북에서 해당 정보를 삭제하세요. 그래야 노트북을 공유할 때 자격 증명이 실수로 노출되지 않습니다. 자세한 안내는 IBM Cloud 계정 설정 및 신뢰할 수 없는 환경에서 서비스 초기화를 참고하세요.
# Load the Qiskit Runtime service
from qiskit_ibm_runtime import QiskitRuntimeService
# Load the Runtime primitive and session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# Syntax for first saving your token. Delete these lines after saving your credentials.
# QiskitRuntimeService.save_account(channel='ibm_quantum_platform',
# instance = '<YOUR_IBM_INSTANCE_CRN>', token='<YOUR-API_KEY>', overwrite=True, set_as_default=True)
# service = QiskitRuntimeService(channel='ibm_quantum_platform')
# Load saved credentials
service = QiskitRuntimeService()
# Use the least busy backend, or uncomment the loading of a specific backend like "ibm_brisbane".
backend = service.least_busy(operational=True, simulator=False, min_num_qubits=127)
# backend = service.backend("ibm_brisbane")
print(backend.name)
ibm_brisbane
아래 셀을 사용하면 노트북 전체에서 시뮬레이터와 실제 하드웨어 사이를 전환할 수 있습니다. 지금 실행해 보세요:
# Load the Aer simulator and generate a noise model based on the currently-selected backend.
from qiskit_aer import AerSimulator
from qiskit_aer.noise import NoiseModel
# Alternatively, load a fake backend with generic properties and define a simulator.
noise_model = NoiseModel.from_backend(backend)
# Define a simulator using Aer, and use it in Sampler.
backend_sim = AerSimulator(noise_model=noise_model)
1단계: 문제를 양자 Circuit과 연산자로 매핑하기
VQE 계산은 특정 결합 거리에서 수소 분자()의 해밀토니안을 정의하는 것으로 시작합니다. 이 해밀토니안은 표준 절차를 통해 분자 시스템에서 생성·매핑된 qubit 연산자 형태로 시스템의 전체 에너지를 표현합니다. 구체적으로는: 1) STO-6G 기저 집합(전자 오비탈을 근사하는 수학 함수들의 특정 모음)을 사용하고, 2) Jordan-Wigner 매핑(전자를 기술하는 페르미온 연산자를 qubit 연산자로 변환하는 기법)을 적용하며, 3) 해밀토니안의 패리티(parity)를 이용한 qubit 감소를 수행하여 문제를 단순화합니다.
앞서 설명한 바와 같이, 계산된 바닥 상태 에너지는 기저 집합 선택과 분자 기하 구조(결합 거리 등)에 크게 의존합니다. 이 특정 구성에서 변환을 적용하면 결과적으로 얻어지는 qubit 해밀토니안은 다음과 같이 간단합니다:
여기서 는 항등 연산자, 는 단일 Qubit에 작용하는 파울리-Z 연산자를 나타냅니다. 계수는 이 특정 결합 거리에서 STO-6G 기저 집합을 사용하여 계산한 적분 값과 적절한 변환으로부터 유도됩니다.
이 해밀토니안이 정의되었으므로, 이제 VQE를 사용하여 바닥 상태 에너지를 계산할 수 있습니다. 계산된 바닥 상태 에너지를 기대값과 비교하는 것이 유용합니다. 단일 고립 수소 원자(H)의 경우 바닥 상태 에너지는 정확히 -0.5 Hartree입니다(상대론적 효과가 없는 경우). 위에서 정의한 특정 qubit 해밀토니안의 정확한 바닥 상태 에너지를 계산하고, 관련된 알려진 값들과 비교해 보겠습니다.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
# Qubit Hamiltonian of the hydrogen atom generated by using STO-3G basis set and parity mapping
Hamiltonian = SparsePauliOp.from_list([("I", -0.2355), ("Z", 0.2355)])
# exact ground state energy of Hamiltonian
A = np.array(Hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print(
"The exact ground state energy of the Hamiltonian is ",
min(eigenvalues).real,
"hartree",
)
h = min(eigenvalues.real)
The exact ground state energy of the Hamiltonian is -0.471 hartree
다음으로, 바닥 상태의 시험 파동 함수 을 준비하기 위한 매개변수화된 양자 Circuit, 즉 앤사츠(ansatz)가 필요합니다. 목표는 에너지 기댓값 을 최소화하는 매개변수 를 찾는 것입니다. 앤사츠의 선택은 매우 중요한데, Circuit이 준비할 수 있는 양자 상태의 집합을 결정하기 때문입니다. "좋은" 앤사츠는 연구 중인 해밀토니안의 실제 바닥 상태에 가까운 상태를 표현할 수 있을 만큼 유연하면서도, 현재 양자 컴퓨터에서 다루기 어려울 만큼 너무 많은 매개변수나 너무 깊은 Circuit을 필요로 하지 않는 것입니다.
여기서는 세 가지 서로 다른 단일 qubit 앤사츠를 시도하여 어느 앤사츠가 더 넓은 "커버리지(coverage)"를 제공하는지 확인해 보겠습니다. 커버리지란 앤사츠 Circuit이 매개변수를 변화시켜 생성할 수 있는 양자 상태의 범위를 의미합니다.
단일 qubit 회전 Gate의 다양한 조합을 기반으로 한 세 가지 앤사츠를 사용합니다:
- 1축 회전 Gate 앤사츠: 이 앤사츠는 단일 축()을 중심으로만 회전합니다. 블로흐 구면에서 이는 특정 원을 따라서만 움직이는 것에 해당합니다. 유연성이 가장 낮으며 제한된 상태 집합만 다룰 수 있습니다.
- 2축 회전 Gate 앤사츠 2개: 이 앤사츠들은 두 개의 서로 다른 축을 중심으로 한 회전을 결합합니다( 및 ). 이를 통해 단일 축 회전에 비해 블로흐 구면의 더 넓은 영역에 도달할 수 있습니다.
세 가지 앤사츠로 얻은 VQE 결과를 비교함으로써, 앤사츠의 유연성과 상태 공간 커버리지가 단순화된 해밀토니안의 실제 바닥 상태 에너지를 찾는 능력에 어떤 영향을 미치는지 알 수 있습니다. 더 유연한 앤사츠는 더 나은 근사값을 찾을 가능성이 있지만, 고전 최적화기가 다루기 더 어려울 수도 있습니다.
from qiskit import QuantumCircuit
from qiskit.circuit import Parameter
from qiskit.quantum_info import Statevector, DensityMatrix, Pauli
theta = Parameter("θ")
phi = Parameter("φ")
lam = Parameter("λ")
ansatz1 = QuantumCircuit(1)
ansatz1.rx(theta, 0)
ansatz2 = QuantumCircuit(1)
ansatz2.rx(theta, 0)
ansatz2.rz(phi, 0)
ansatz3 = QuantumCircuit(1)
ansatz3.rx(theta, 0)
ansatz3.rz(phi, 0)
ansatz3.rx(lam, 0)
<qiskit.circuit.instructionset.InstructionSet at 0x1059def80>
이제 각 매개변수에 대해 5000개의 난수를 생성하고, 세 가지 앤사츠가 이 무작위 매개변수로 생성한 양자 상태의 분포를 플롯으로 나타내 보겠습니다. 이 매개변수들은 구면 표면의 서로 다른 축을 중심으로 한 회전처럼 생각할 수 있습니다. 양자 상태의 분포를 확인하기 위해 블로흐 구면(Bloch sphere)을 사용합니다. 블로흐 구면은 단일 Qubit의 상태를 보여주는 3차원 구면입니다. 구면 위의 모든 점은 Qubit의 가능한 상태를 나타내며, 북극과 남극은 고전적인 "0"과 "1"에 해당하지만, Qubit은 그 사이 어느 곳에도 있을 수 있어 중첩과 같은 특별한 양자 특성을 보입니다. 먼저 3D 블로흐 구면을 플롯하고 5000개의 무작위 매개변수를 준비하는 데 필요한 함수를 준비합니다.
import matplotlib.pyplot as plt
def plot_bloch(bloch_vectors):
# Extract X, Y, Z coordinates for 3D projection
X_coords = bloch_vectors[:, 0]
Z_coords = bloch_vectors[:, 2]
# Compute Y coordinates from X and Z to approximate the full Bloch sphere projection
Y_coords = bloch_vectors[:, 1]
# Create 3D plot
fig = plt.figure(figsize=(8, 8))
ax = fig.add_subplot(111, projection="3d")
ax.scatter(X_coords, Y_coords, Z_coords, color="blue", alpha=0.6)
# Labels and title
ax.set_xlabel("X")
ax.set_ylabel("Y")
ax.set_zlabel("Z")
ax.set_title("Parameterized 1-Qubit Circuit on 3D Bloch Sphere")
# Set axis limits and make them equal
ax.set_xlim([-1, 1])
ax.set_ylim([-1, 1])
ax.set_zlim([-1, 1])
# Ensure equal aspect ratio for all axes
ax.set_box_aspect([1, 1, 1]) # Equal scaling for x, y, z axes
# Show grid
ax.grid(True)
plt.show()
num_samples = 5000 # Number of random states
theta_vals = np.random.uniform(0, 2 * np.pi, num_samples)
phi_vals = np.random.uniform(0, 2 * np.pi, num_samples)
lam_vals = np.random.uniform(0, 2 * np.pi, num_samples)
첫 번째 앤사츠가 어떻게 작동하는지 살펴보겠습니다.
# List to store Bloch Sphere XZ coordinates
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create a circuit and bind parameters
qc = ansatz1
bound_qc = qc.assign_parameters({theta: theta_vals[i]}) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to a numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
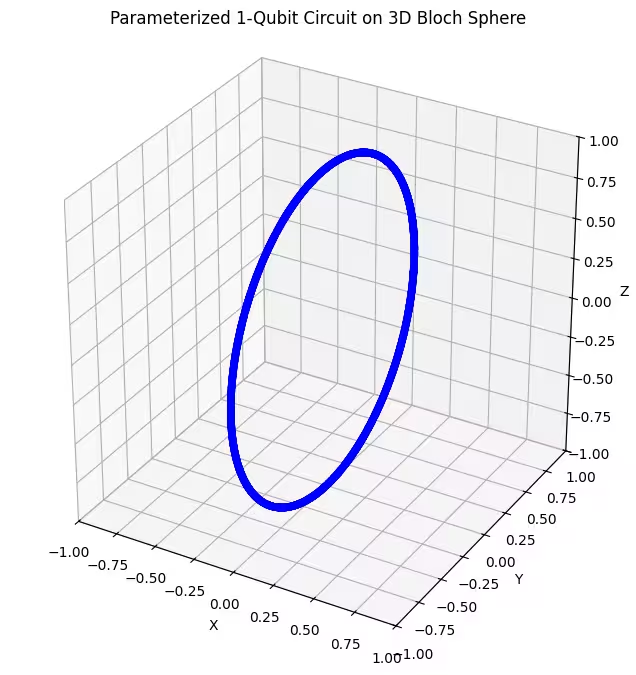
첫 번째 앤사츠는 블로흐 구면에서 고리 모양의 양자 상태 분포를 반환하는 것을 볼 수 있습니다. 이는 앤사츠에 단 하나의 회전 매개변수만 주었기 때문에 당연한 결과입니다. 따라서 한 축을 중심으로 회전한 상태만 생성할 수 있습니다. 지점에서 시작하여 한 축을 중심으로 회전하면 항상 고리가 생성됩니다. 이제 두 개의 직교 회전 Gate, 즉 Rx와 Rz를 사용하는 두 번째 앤사츠를 확인해 보겠습니다.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz2
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i]}
) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
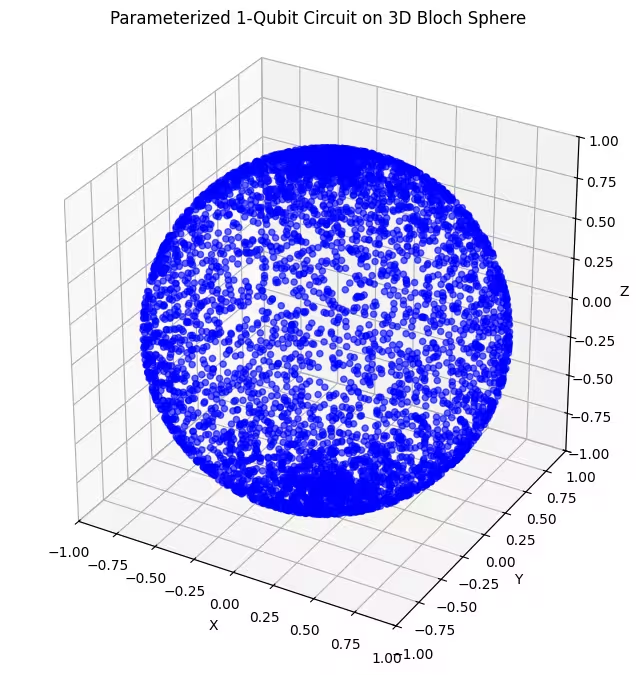
두 번째 앤사츠는 블로흐 구면의 더 넓은 영역을 커버하지만, 점들이 극점 근처에서는 더 밀집되고 적도 근처에서는 더 분산되어 있음을 알 수 있습니다. 이제 마지막 앤사츠를 확인할 차례입니다.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz3
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i], lam: lam_vals[i]}
)
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
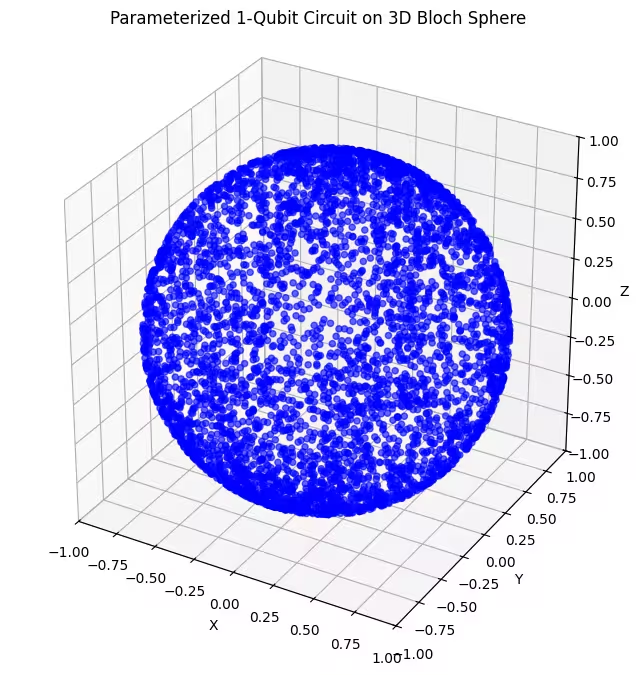
마지막 앤사츠가 생성하는 양자 상태가 더 고르게 분포되어 있음을 볼 수 있습니다.
앞서 언급했듯이, 가장 좋은 방법은 찾고자 하는 바닥 상태에 대한 지식을 바탕으로 그 바닥 상태에 가까운 상태를 효과적으로 탐색할 수 있는 앤사츠를 선택하는 것입니다. 예를 들어, 바닥 상태가 극점 근처에 있다는 것을 알고 있다면 앤사츠 2를 선택할 수 있습니다. 간단함을 위해, 블로흐 구면 전체를 균일하게 탐색하는 앤사츠 3을 사용하겠습니다.
앤사츠를 선택했으니, 이제 Circuit을 그려 보겠습니다.
# Pre-defined ansatz circuit and operator class for Hamiltonian
ansatz = ansatz3
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.draw("mpl", style="iqp")
This circuit has 3 parameters

Step 2: 목표 하드웨어에 맞게 최적화하기
실제 양자 컴퓨터에서 계산을 실행할 때는 양자 회로의 논리만 고려하는 것이 아닙니다. 특정 양자 컴퓨터가 수행할 수 있는 연산이 무엇인지, 그리고 사용 중인 Qubit이 양자 컴퓨터의 어느 위치에 있는지도 고려해야 합니다. 서로 인접해 있나요? 멀리 떨어져 있나요? 따라서 다음 단계는 사용할 양자 컴퓨터에 자연스러운 Gate를 사용하여 회로를 재작성하고, qubit 레이아웃을 반영하는 것입니다. 이는 transpilation(트랜스파일)을 통해 수행할 수 있습니다. 이 과정을 거치면 단순했던 앤사츠가 다른 Gate 집합으로 변환되고, 추상적인 Qubit이 실제 양자 컴퓨터의 물리적 Qubit에 매핑되는 것을 확인할 수 있습니다.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
config = backend.configuration()
print("Backend: {config.backend_name}")
print("Native gates: ", config.supported_instructions, ",")
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")
Backend: {config.backend_name}
Native gates: ['ecr', 'id', 'delay', 'measure', 'reset', 'rz', 'sx', 'x'] ,

ansatz의 rx, rz Gate가 Backend의 네이티브 Gate인 rz, sx Gate의 연속으로 변환된 것을 확인할 수 있습니다. 또한 q0가 다섯 번째 물리적 Qubit에 매핑된 것도 볼 수 있습니다. 다음 코드와 같이 이러한 변경 사항에 맞춰 해밀토니안도 매핑해야 합니다:
Hamiltonian_isa = Hamiltonian.apply_layout(layout=ansatz_isa.layout)
Step 3: 목표 하드웨어에서 실행하기
이제 실제 QPU에서 VQE를 실행할 차례입니다. 이를 위해 먼저 최적화 과정에서 사용할 비용 함수(cost function)가 필요합니다. 이 함수는 ansatz가 생성한 양자 상태로 해밀토니안의 기댓값을 평가합니다. 걱정하지 마세요! 모든 것을 직접 코딩할 필요는 없습니다. 이를 위한 함수를 미리 준비해 두었으니, 아래 셀을 실행하기만 하면 됩니다.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
다음으로, ansatz의 초기 매개변수와 최적화 과정을 준비합니다. 모두 0으로 설정하거나 랜덤 값을 사용할 수 있습니다. 아래에 초기 매개변수를 선택해 두었지만, 셀의 줄을 주석 처리하거나 해제하여 에서 사이의 균등 분포에서 매개변수를 무작위로 샘플링해도 좋습니다.
# x0 = np.random.uniform(0, 2*pi, 3)
x0 = [1, 1, 0]
# QPU Est. 2min for ibm_brisbane
from scipy.optimize import minimize
from qiskit_ibm_runtime import Batch
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, Hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 10, "tol": 0.01},
)
batch.close()
Iters. done: 1 [Current cost: -0.3361517318448143]
Iters. done: 2 [Current cost: -0.4682546422099432]
Iters. done: 3 [Current cost: -0.38985802144149584]
Iters. done: 4 [Current cost: -0.38319217316749354]
Iters. done: 5 [Current cost: -0.4628720756579032]
Iters. done: 6 [Current cost: -0.4683301936226905]
Iters. done: 7 [Current cost: -0.45480498699294747]
Iters. done: 8 [Current cost: -0.4690533242050814]
Iters. done: 9 [Current cost: -0.465867415110354]
Iters. done: 10 [Current cost: -0.4606882723137227]
h_vqe = res.fun
print("The reference ground state energy is ", min(eigenvalues))
print("The computed ground state energy is ", h_vqe)
The reference ground state energy is (-0.471+0j)
The computed ground state energy is -0.4690533242050814
축하합니다! 첫 번째 양자 화학 실험을 성공적으로 마쳤습니다. 해밀토니안의 정확한 바닥 상태 에너지와 우리가 구한 값 사이에 차이가 있지만, 기본 오류 완화 기법(읽기 오류를 보정)을 사용했기 때문에 그 차이는 미미합니다. 아주 좋은 시작입니다!
참고: resilience_level을 사용하여 오류 완화 수준을 설정하면 더 나은 결과를 얻을 수 있습니다. 기본값은 1이며, 더 높은 값을 설정하면 QPU 시간은 더 소요되지만 더 나은 결과를 반환할 수 있습니다.
Step 4: 후처리하기
이제 고전 최적화기가 어떻게 동작했는지 살펴볼 차례입니다. 아래 셀을 실행하여 수렴 패턴을 확인해 보세요.
fig, ax = plt.subplots()
x = np.linspace(0, 10, 10)
# Define the constant function
y_constant = np.full_like(x, h)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

상당히 좋은 초기값으로 시작하여 단 10단계만에 좋은 최종값을 얻었습니다. 크고 작은 피크가 보이는데, 이것은 COBYLA 최적화기의 전형적인 특징입니다. COBYLA는 마치 경관을 볼 수 없는 것처럼 공간을 탐색하면서 각 측정마다 탐색 보폭을 조정합니다.
이해도 확인하기
어떤 점을 관찰했나요? 이론값에 더 가깝거나 해밀토니안의 정확한 바닥 상태 에너지에 더 근접한 결과를 얻기 위해 위 과정에서 어느 부분을 개선할 수 있을까요? 고려해야 할 사항은 무엇인가요?
Answer
먼저 고려할 것은 분자의 해밀토니안을 계산하는 데 사용되는 기저 함수(basis) 집합의 변경입니다. 앞서 언급했듯이, H 원자의 바닥 상태 에너지는 잘 알려진 바와 같이 -0.5 Hartree이지만, 선택한 STO-6G 기저는 이 값을 정확하게 도출하기에 충분하지 않습니다.
더 복잡한 기저를 선택하면 해밀토니안에서 사용되는 qubit 수가 늘어나므로, 화학 문제에 더 복잡하고 적합한 앤사츠를 선택해야 합니다.
다음으로 최적화해야 할 것은 QPU에서의 노이즈 관리입니다. 더 고급 오류 완화 기법을 사용하면 더 나은 결과를 얻을 수 있지만, 시간이 더 오래 걸릴 수 있습니다. 또한 shot_number가 결과에 어떤 영향을 미치는지도 고려해 보세요.
마지막으로, 다른 최적화기를 시도하여 더 나은 수렴 성능을 달성할 수도 있습니다.
VQE를 이용한 수소 분자 바닥 상태 에너지 계산
원자를 이용해 VQE의 전체적인 과정을 살펴봤으니, 이제 분자의 바닥 상태 에너지를 좀 더 빠르게 계산해 보겠습니다.
Step 1: 문제를 양자 회로와 연산자로 매핑하기
여기서도 STO-6G 기저와 Jordan-Wigner 변환을 사용하고, 해밀토니안의 대칭성을 이용한 qubit 수 감소를 적용한 1-Qubit 해밀토니안을 제공합니다. 두 수소 원자 사이의 원자 간 거리는 0.735 를 사용했음에 유의하세요.
단일 수소 원자()의 계산과 달리, 수소 분자()의 바닥 상태를 계산하려면 전자 오비탈과 관련된 에너지 외에도 두 수소 원자의 핵 사이에 작용하는 반발력을 함께 고려해야 합니다. 이 단계에서는 이 값을 상수로 제공하며, 실제 계산은 연습 문제에서 다루겠습니다.
h2_hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
# exact ground state energy of hamiltonian
nuclear_repulsion = 0.71997
A = np.array(h2_hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print("Electronic ground state energy (Hartree): ", min(eigenvalues).real)
print("Nuclear repulsion energy (Hartree): ", nuclear_repulsion)
print(
"Total ground state energy (Hartree): ", min(eigenvalues).real + nuclear_repulsion
)
h2 = min(eigenvalues).real + nuclear_repulsion
Electronic ground state energy (Hartree): -1.8659468547627318
Nuclear repulsion energy (Hartree): 0.71997
Total ground state energy (Hartree): -1.1459768547627318
Step 2: 대상 하드웨어를 위한 최적화
이전 VQE와 해밀토니안에 사용된 qubit 수가 실행에 사용할 Backend와 동일하므로, 기존 ansatz와 그 최적화된 형태를 그대로 사용합니다.
h2_hamiltonian_isa = h2_hamiltonian.apply_layout(layout=ansatz_isa.layout)
Step 3: 대상 하드웨어에서 실행하기
이제 실제 QPU에서 계산을 수행할 차례입니다. 거의 모든 것이 동일하지만, 해밀토니안에 맞는 적절한 초기 파라미터를 사용합니다. 또한 반복 단계에서 QPU 내의 ansatz에 대한 해밀토니안 기댓값을 계산하는 데 사용되는 Estimator의 일부 설정이 이전 계산과 약간 다르게 지정됩니다. 이 변경 사항에 대해서는 연습 문제에서 더 자세히 다루겠습니다.
x0 = [2, 0, 0]
# QPU time 4min for ibm_brisbane
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, h2_hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 15},
)
batch.close()
Iters. done: 1 [Current cost: -0.710621837568328]
Iters. done: 2 [Current cost: -0.2603208441168329]
Iters. done: 3 [Current cost: -0.25548711201326424]
Iters. done: 4 [Current cost: -0.581129450619904]
Iters. done: 5 [Current cost: -1.722920997605439]
Iters. done: 6 [Current cost: -1.6633324849371915]
Iters. done: 7 [Current cost: -1.8066989598929164]
Iters. done: 8 [Current cost: -1.8051093803839542]
Iters. done: 9 [Current cost: -1.802692217571555]
Iters. done: 10 [Current cost: -1.8233585485263144]
Iters. done: 11 [Current cost: -1.6904116652617205]
Iters. done: 12 [Current cost: -1.8245120321245392]
Iters. done: 13 [Current cost: -1.6837021361383608]
Iters. done: 14 [Current cost: -1.8166632606115467]
Iters. done: 15 [Current cost: -1.863446212658907]
h2_vqe = res.fun + nuclear_repulsion
print(
"The reference ground state energy is ", min(eigenvalues).real + nuclear_repulsion
)
print("The computed ground state energy is ", h2_vqe)
The reference ground state energy is -1.1459768547627318
The computed ground state energy is -1.143476212658907
이론적으로 VQE는 진정한 바닥 상태 에너지의 상한(upper bound)을 제공하지만, 실제 양자 하드웨어나 잡음이 있는 시뮬레이션 환경에서의 구현, 그리고 해밀토니안 준비 과정에서의 근사(기저 집합이나 qubit 수 감소 등)는 측정된 에너지가 이론적 정확값 또는 특정 수치 기준보다 약간 낮게 나오는 오류를 유발할 수 있습니다. 약간의 오차가 있지만, 특히 적은 반복 횟수를 고려하면 결과는 만족스러운 수준입니다. 이제 최적화기가 어떻게 동작했는지 살펴보며 이번 VQE 계산을 마무리해 봅시다.
Step 4: 후처리
fig, ax = plt.subplots()
x = np.linspace(0, 5, 15)
# Define the constant function
y_constant = np.full_like(x, min(eigenvalues))
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

이해도 확인
상수로 포함시켰던 분자의 핵 반발 에너지(0.71997 Hartree)를 직접 계산해 봅시다.

쿨롱 법칙과 원자 단위계를 사용하여 값이 Hartree 단위로 나오도록 계산해 보세요.
Answer
두 수소 핵은 모두 양전하를 띠므로 정전기적 힘에 의해 서로 반발합니다. 이 반발력은 쿨롱 법칙으로 설명됩니다.
여기서 는 양성자의 전하, 은 진공 유전율, 은 두 핵 사이의 거리(미터 또는 보어 반지름 단위, 에너지 단위는 줄(J))입니다.
이 에너지를 Hartree 단위로 계산하려면 위 식을 원자 단위계(AU)로 변환해야 합니다. AU에서는 , 이고, 보어 반지름()이 1이 되어 AU의 기본 길이 척도가 됩니다. 이 단순화를 적용하면 쿨롱 법칙은 다음과 같이 줄어듭니다.
여기서 은 보어 반지름() 단위로 측정해야 합니다.
주어진 핵 간 거리를 에서 으로 변환하려면 다음 변환 관계를 이용합니다.
따라서 는 이 됩니다.
그러므로 주어진 의 핵 반발 에너지는
의 반응 에너지 계산
지금까지 얻은 결과를 활용해 봅시다! 여러분은 VQE(변분 양자 고유값 분해기)를 이용해 원자와 분자의 바닥 상태 에너지를 각각 계산했습니다. 이제 남은 것은 계산된 값을 사용하여 과정의 반응 에너지를 구하는 것입니다.
반응 에너지란 물질이 반응하여 새로운 물질을 생성할 때 발생하는 에너지 변화를 말합니다. 무언가를 만드는 상황을 상상해 보세요. 어떤 경우에는 에너지를 투입해야 하고(블록을 쌓는 것처럼), 어떤 경우에는 에너지가 방출되기도 합니다(공이 언덕을 굴러 내려가는 것처럼). 화학에서 반응은 에너지를 흡수하는 흡열 반응이거나 에너지를 방출하는 발열 반응입니다.
과정의 반응 에너지는 다음 공식으로 계산할 수 있습니다.
아래 셀을 실행하여 이를 시각적으로 확인해 봅시다. 여기서는 각 해밀토니안의 정확한 바닥 상태 값을 사용하고, 정확한 해와 VQE 결과의 반응 에너지를 비교합니다.
# Theoretical values
E_H_theo = h.real
E_H2_theo = h2
# Experimental values
E_H_exp = h_vqe
E_H2_exp = h2_vqe
# Calculate reaction energies
E_reaction_theo = E_H2_theo - (2 * E_H_theo)
E_reaction_exp = E_H2_exp - (2 * E_H_exp)
# Set up the plot
fig, ax = plt.subplots(figsize=(8, 6))
ax.set_xlim(0, 3)
ax.set_ylim(-1.16, -0.93) # Adjust y-axis range to highlight differences
ax.set_xticks([])
ax.set_ylabel("Energy (Hartree)")
ax.set_title("H + H → H₂ Reaction Energy Diagram")
# Plot theoretical energy levels
ax.hlines(
y=2 * E_H_theo, xmin=0.5, xmax=1.3, linewidth=2, color="r", label="2H (Exact)"
)
ax.hlines(y=E_H2_theo, xmin=1.3, xmax=2, linewidth=2, color="b", label="H₂ (Exact)")
# Plot experimental energy levels
ax.hlines(
y=2 * E_H_exp,
xmin=0.5,
xmax=1.5,
linewidth=2,
color="r",
linestyle="dashed",
label="2H (VQE)",
)
ax.hlines(
y=E_H2_exp,
xmin=1.5,
xmax=2.5,
linewidth=2,
color="b",
linestyle="dashed",
label="H₂ (VQE)",
)
# Add labels
ax.text(
1,
2 * E_H_theo,
f"2H: {2*E_H_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
2,
E_H2_theo,
f"H₂: {E_H2_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
1,
2 * E_H_exp,
f"2H_VQE: {2*E_H_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
ax.text(
2,
E_H2_exp,
f"H₂_VQE: {E_H2_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
# Add arrows for reaction energy with ΔE label in the middle
mid_y_theo = (2 * E_H_theo + E_H2_theo) / 2
mid_y_exp = (2 * E_H_exp + E_H2_exp) / 2
ax.annotate(
"",
xy=(1.3, E_H2_theo),
xytext=(1.3, 2 * E_H_theo),
arrowprops=dict(arrowstyle="<->", color="g"),
)
ax.text(
1.35, mid_y_theo, f"ΔE: {E_reaction_theo:.4f}", color="g", verticalalignment="top"
)
ax.annotate(
"",
xy=(1.5, E_H2_exp),
xytext=(1.5, 2 * E_H_exp),
arrowprops=dict(arrowstyle="<->", color="g", linestyle="dashed"),
)
ax.text(
1.55,
mid_y_exp,
f"ΔE_VQE: {E_reaction_exp:.4f}",
color="g",
verticalalignment="center",
)
# Add legend
ax.legend()
plt.show()

그림에서 볼 수 있듯이, 다소 오차가 있지만 해밀토니안의 정확한 바닥 상태 에너지와 VQE 결과를 이용해 계산한 반응 에너지는 모두 -0.2 Hartree에 근접하여 유사한 결과를 보여줍니다.
여기서 주목할 점은 이 과정의 반응 에너지가 음수 값을 가진다는 것입니다. 이는 이 과정을 통해 에너지가 방출되며, 생성된 분자가 두 개의 단일 원자보다 더 낮은 에너지 상태에 있음을 의미합니다.
- 결론
지금까지 배운 내용을 정리해 봅시다.
먼저 양자 화학 문제를 풀기 위해 필요한 두 가지 중요한 근사 기법인 변분 원리와 기저 집합 선택을 살펴봤습니다. 이 두 가지는 모두 VQE의 근본적인 요소입니다. 단순 조화 진동자의 바닥 상태 에너지를 직접 계산하며 변분 원리를 탐구했습니다.
다음으로 양자 시스템의 바닥 상태 에너지를 계산하는 데 널리 사용되는 알고리즘인 VQE를 살펴봤습니다. 수소 원자()와 수소 분자()의 바닥 상태 에너지를 계산하는 코드를 실행했습니다. 특히, 해당 시스템에 적합한 분자 해밀토니안을 구하고 이를 양자 컴퓨터에서 실행 가능한 형태로 변환하는 것이 필요하다는 것을 배웠습니다. 또한 VQE에서 시험 양자 상태를 준비하기 위해 매개변수화된 양자 Circuit인 앤사츠(ansatz)가 필요하며, 적절한 앤사츠 Circuit 구조를 선택하는 것이 중요하다는 점도 살펴봤습니다. 아울러 VQE는 고전 컴퓨터를 이용한 반복적인 최적화 과정에 의존하여 양자 Circuit이 최저 에너지 상태를 찾도록 유도하며, 이 과정이 어떻게 수렴되는지도 확인했습니다.
마지막으로 VQE를 통해 계산된 와 의 바닥 상태 에너지를 사용하여 과정의 반응 에너지를 계산했습니다.
VQE는 강력한 근거리 양자 알고리즘이지만 그 한계도 인식하는 것이 중요합니다. VQE의 성능은 앤사츠 선택에 크게 의존하는데, 더 크고 복잡한 분자의 경우 실제 바닥 상태를 정확하게 표현할 수 있으면서도 효율적으로 준비 가능한 앤사츠를 찾는 것이 어려워집니다. 또한 현재의 양자 하드웨어는 노이즈에 취약하여, 특히 더 깊은 Circuit이나 더 많은 수의 Qubit을 다루는 경우 VQE 결과의 정확도에 영향을 줄 수 있습니다. 이러한 한계에도 불구하고, VQE는 기초 알고리즘으로서의 역할을 하며, 현재 진행 중인 연구들은 근거리 양자 컴퓨터에서 양자 화학의 가능성을 확장하기 위한 더 정교한 변분 방법과 오류 완화 기법을 탐색하고 있습니다. 예를 들어, SQD(샘플 기반 양자 대각화)와 같은 알고리즘이 개발 중인데, 이는 양자 Circuit에서 얻은 샘플과 부분 공간에서의 고전적 대각화를 결합하여 에너지 추정을 개선하고, 특히 측정 효율성과 노이즈 강건성 측면에서 VQE의 한계를 해결하고자 합니다.
복습 및 질문
핵심 개념:
- 변분 양자 알고리즘은 고전 컴퓨터와 양자 컴퓨터가 협력하여 문제를 해결하는 컴퓨팅 패러다임입니다.
- VQE에서는 시스템의 해밀토니안으로 시작하여 이를 양자 컴퓨터에서 실행할 수 있도록 Qubit에 매핑합니다. 매개변수화된 양자 Circuit인 앤사츠를 선택하고, 최저 에너지 값에 도달할 때까지 앤사츠의 매개변수를 변경하면서 반복적으로 측정합니다. 매개변수 공간 탐색은 고전 최적화기를 사용하여 수행됩니다. 좋은 결과를 얻으려면 적절한 앤사츠와 최적화기를 선택하는 것이 필요합니다.
- 반응 에너지는 화학 반응에서의 총 에너지 변화로, 반응물과 생성물의 에너지 차이로 결정됩니다.
참/거짓
- 변분 원리는 임의의 시험 파동함수에 대한 에너지의 기댓값이 항상 실제 바닥 상태 에너지보다 크거나 같다고 말합니다.
- 기저 집합은 양자 파동함수를 근사하는 데 사용되는 함수들의 모음입니다.
- VQE는 주어진 해밀토니안에 대해 슈뢰딩거 방정식을 정확하게 풀기 위한 양자 알고리즘입니다.
- VQE에서는 매개변수화된 양자 Circuit(앤사츠)을 사용하여 시험 파동함수를 준비합니다.
- VQE에서 최적화기의 선택(예: COBYLA, SPSA, ADAM)은 결과의 품질에 영향을 미치지 않습니다.
- Qiskit의
Estimator는 VQE에서 해밀토니안의 기댓값을 직접 계산하는 데 사용됩니다.
객관식 문제:
- VQE에서 해밀토니안의 목적은 무엇인가요?
- A) 무작위 양자 상태를 생성하기 위해
- B) 양자 상태의 에너지를 결정하기 위해
- C) 양자 Circuit을 최적화하기 위해
- D) 얽힘을 생성하기 위해
- VQE 알고리즘의 주요 목표는 무엇인가요?
- A) 해밀토니안의 바닥 상태 에너지를 찾기 위해
- B) qubit 간 얽힘을 생성하기 위해
- C) 그로버 탐색을 수행하기 위해
- D) RSA 암호화를 해독하기 위해
- 이 노트북에서 앤사츠를 비교하기 위해 생성되는 양자 상태는 몇 개인가요?
- A) 100
- B) 1000
- C) 5000
- D) 10,000
- VQE에 고전 최적화기가 필요한 이유는 무엇인가요?
- A) 양자 측정을 수행하기 위해
- B) 에너지를 최소화하도록 앤사츠 매개변수를 업데이트하기 위해
- C) Qubit을 얽히게 하기 위해
- D) 양자 무작위성을 생성하기 위해
- 앤사츠가 매개변수화되도록 설계되는 이유는 무엇인가요?
- A) 양자 상태 준비를 가능하게 하기 위해
- B) 넓은 양자 상태 공간을 탐색할 수 있게 하기 위해
- C) Circuit 복잡성을 줄이기 위해
- D) 고유값을 직접 측정하기 위해
- 좋은 앤사츠를 선택하는 것에 대해 가장 올바른 설명은 무엇인가요?
- A) 앤사츠는 블로흐 구 위에 균등하게 분포된 상태를 생성해야 하며, 그렇지 않으면 실패합니다.
- B) 앤사츠는 바닥 상태에 가까운 상태를 생성할 수 있도록 시스템에 맞게 설계되어야 합니다.
- C) 앤사츠는 변분 매개변수를 사용하여 무작위 상태를 생성해야 합니다.
- D) 더 좋은 앤사츠는 항상 더 많은 변분 매개변수를 가집니다.
(Optional) 부록: Ansatz 복잡도에 따른 최적화기 오버헤드
VQE는 잘 알려진 여러 가지 과제[ref 6]에 직면해 있으며, 다음 내용은 앞서 학습한 내용과 관련이 있습니다.
- Ansatz 선택의 어려움
적절한 변분 Ansatz를 선택하는 데는 본질적인 어려움이 있습니다. 화학에서 영감을 받은 Ansatz(예: UCCSD)는 물리적 정확도를 제공하지만 깊은 Circuit을 필요로 하는 반면, 하드웨어 효율적인 Ansatz는 더 얕은 Circuit을 가지지만 물리적 해석 가능성이 부족할 수 있습니다. 또한 많은 Ansatz는 정확도 향상에 거의 기여하지 않으면서 최적화 난이도를 크게 높이는 과도한 변분 매개변수를 도입합니다.
- 최적화의 어려움
VQE의 최적화 지형에는 기울기가 지수적으로 소실되는 영역(barren plateau)이 존재할 수 있어, 고전 최적화기가 변분 매개변수를 효율적으로 업데이트하기 어렵게 만듭니다. 이를 위해 연구자들은 기울기 기반 및 기울기 없는 방식 등 다양한 유형의 최적화기를 시도해 왔지만, 두 방식 모두 과제를 안고 있습니다. 기울기 기반 최적화기는 barren plateau 문제를 겪고, 기울기 없는 방법은 많은 수의 함수 평가를 필요로 합니다.
- 최적화기 오버헤드
또 다른 잘 알려진 과제는 최적화기 오버헤드로, 이는 문제의 규모와 관련이 있습니다. VQE에 필요한 양자 Circuit은 문제 크기가 증가함에 따라 깊이와 복잡도가 커지며, 이는 일반적으로 최적화해야 할 매개변수의 수도 증가시킵니다. 매개변수 수가 늘어날수록 최적화 과정은 다루기 어려워지고, 수렴이 느려지며 최적해를 찾는 데 어려움이 생깁니다.
여기서는 두 가지 유형의 Ansatz를 사용하여 분자에 VQE를 적용함으로써 이러한 과제들을 살펴보겠습니다.
(참고: 이 과정은 더 많은 QPU 시간이 소요될 수 있으므로, 시간이 충분하지 않다면 시뮬레이터를 사용해도 됩니다.)
from qiskit.circuit import ParameterVector
num_iter = 4
alpha = ParameterVector("alpha", 3)
beta = ParameterVector("beta", 3 * num_iter)
# step1: Map problem to quantum circuits and operators
hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
ansatz_1 = ansatz3
ansatz_2 = QuantumCircuit(1)
for i in range(num_iter):
ansatz_2.rx(beta[i * 3 + 0], 0)
ansatz_2.rz(beta[i * 3 + 1], 0)
ansatz_2.rx(beta[i * 3 + 2], 0)
ansatz_1.draw("mpl")
ansatz_2.draw("mpl")

# Step 2: Optimize for target hardware
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa_1 = pm.run(ansatz_1)
ansatz_isa_2 = pm.run(ansatz_2)
hamiltonian_isa_1 = hamiltonian.apply_layout(layout=ansatz_isa_1.layout)
hamiltonian_isa_2 = hamiltonian.apply_layout(layout=ansatz_isa_2.layout)
이제 모든 값이 1로 설정된 초기 포인트와 최대 20 스텝으로 VQE를 실행하고, 두 실행의 수렴 과정을 비교해 보겠습니다.
# QPU time 3m 40s for ibm_brisbane
# Step 3: Execute on target hardware
from scipy.optimize import minimize
x0 = np.ones(ansatz_1.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_1, hamiltonian_isa_1, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.8782202668652658]
Iters. done: 2 [Current cost: -0.43473160695469165]
Iters. done: 3 [Current cost: -0.4076372093159749]
Iters. done: 4 [Current cost: -1.3587839859772106]
Iters. done: 5 [Current cost: -1.774529906754082]
Iters. done: 6 [Current cost: -1.541934983115727]
Iters. done: 7 [Current cost: -1.2732403113465345]
Iters. done: 8 [Current cost: -1.820842221085785]
Iters. done: 9 [Current cost: -1.8065762857059005]
Iters. done: 10 [Current cost: -1.8126394095981146]
Iters. done: 11 [Current cost: -1.8205831886180421]
Iters. done: 12 [Current cost: -1.8086715778994924]
Iters. done: 13 [Current cost: -1.8307676638629322]
Iters. done: 14 [Current cost: -1.8177328827556327]
Iters. done: 15 [Current cost: -1.8179426218088064]
Iters. done: 16 [Current cost: -1.8109239667991088]
Iters. done: 17 [Current cost: -1.824271872489647]
Iters. done: 18 [Current cost: -1.813167587671394]
Iters. done: 19 [Current cost: -1.824647343397313]
Iters. done: 20 [Current cost: -1.8219785311686143]
# Save Cost_history as a new list
ansatz_1_history = cost_history_dict["cost_history"]
# QPU time 3m 40s for ibm_brisbane
x0 = np.ones(ansatz_2.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_2, hamiltonian_isa_2, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.738191173881188]
Iters. done: 2 [Current cost: -0.42636037194506304]
Iters. done: 3 [Current cost: -1.3503788613797374]
Iters. done: 4 [Current cost: -0.9109204349776897]
Iters. done: 5 [Current cost: -0.9060873157510835]
Iters. done: 6 [Current cost: -0.7735065414083984]
Iters. done: 7 [Current cost: -1.586889197437709]
Iters. done: 8 [Current cost: -1.659215191584943]
Iters. done: 9 [Current cost: -1.245445981794618]
Iters. done: 10 [Current cost: -1.1608385766138023]
Iters. done: 11 [Current cost: -1.1551733876027737]
Iters. done: 12 [Current cost: -1.8143337768286332]
Iters. done: 13 [Current cost: -1.2510951563756598]
Iters. done: 14 [Current cost: -1.6918311531865413]
Iters. done: 15 [Current cost: -1.8163783305531838]
Iters. done: 16 [Current cost: -1.8434877732947152]
Iters. done: 17 [Current cost: -1.8461898233304472]
Iters. done: 18 [Current cost: -1.0346471214915485]
Iters. done: 19 [Current cost: -1.8322518854150687]
Iters. done: 20 [Current cost: -1.717144678705999]
ansatz_2_history = cost_history_dict["cost_history"]
fig, ax = plt.subplots()
# Define the constant function)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_1_history,
label="Ansatz with 3 parameters",
)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_2_history,
label="Ansatz with 12 parameters",
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
plt.legend()
plt.draw()

위 그래프는 변수가 더 많은 Ansatz의 최적화 과정이 안정적인 수렴에 도달하는 데 더 많은 시간이 걸린다는 것을 명확히 보여줍니다.
단순한 단일 qubit Circuit과 간단한 Ansatz에 의존하는 것과 달리, 더 큰 양자 Circuit과 복잡한 구조의 Ansatz가 필요할 때 최적화의 복잡성이 증가합니다. 이는 VQE의 잘 알려진 과제인 최적화기 오버헤드를 잘 보여줍니다.
연구자들은 양자 컴퓨터를 화학 문제에 활용할 수 있는 다양한 고급 방법론을 지속적으로 개발하고 있습니다. IBM Quantum Learning에서 다양한 교육 자료를 이용할 수 있습니다.
참고문헌
- [ref 1 ] Richard P. Feynman, Simulating Physics with Computers, International Journal of Theoretical Physics, 1982.
- [ref 2] Marov, M.Y. (2015). The Structure of the Universe. In: The Fundamentals of Modern Astrophysics. Springer, New York, NY.
- [ref 3] How to solve difficult chemical engineering problems with quantum computing, IBM Research Blog, 2023.
- [ref 4] Y. Cao, J. Romero and A. Aspuru-Guzik, "Potential of quantum computing for drug discovery," in IBM Journal of Research and Development, vol. 62, no. 6, pp. 6:1-6:20, 1 Nov.-Dec. 2018
- [ref 5] Present State of Molecular Structure Calculation, REv. Mod. Phys. 32, 170, 1960
- [ref 6] Fedorov, D.A., Peng, B., Govind, N. et al. VQE method: a short survey and recent developments. Mater Theory 6, 2 (2022)