변분 양자 고유값 분해기 (VQE)
이 단원에서는 변분 양자 고유값 분해기를 소개하고, 양자 컴퓨팅의 기초 알고리즘으로서의 중요성을 설명하며, 장단점도 살펴봅니다. 보조 기법 없이 VQE 단독으로는 현대의 유틸리티 규모 양자 계산에 충분하지 않을 가능성이 높습니다. 그럼에도 VQE는 고전-양자 하이브리드 방식의 원형으로서 중요하며, 더 발전된 많은 알고리즘의 토대가 됩니다.
이 영상은 VQE와 효율성에 영향을 미치는 요인들에 대한 개요를 제공합니다. 아래 본문에서는 더 자세한 내용을 다루고 Qiskit을 이용한 VQE 구현을 소개합니다.
1. VQE란 무엇인가요?
변분 양자 고유값 분해기는 고전 컴퓨팅과 양자 컴퓨팅을 함께 사용하여 작업을 수행하는 알고리즘입니다. VQE 계산에는 크게 네 가지 구성 요소가 있습니다.
- 연산자: 최적화하고자 하는 시스템의 특성을 나타내는 연산자로, 흔히 라고 표기하는 해밀토니안입니다. 다시 말해, 최솟값 고유값에 해당하는 이 연산자의 고유벡터를 찾는 것이 목표입니다. 이 고유벡터를 흔히 "바닥 상태"라고 부릅니다.
- "ansatz" (독일어로 "접근법"): 구하려는 고유벡터를 근사하는 양자 상태를 준비하는 양자 Circuit입니다. 실제로 ansatz는 양자 Circuit의 집합으로, 일부 Gate에 변분 매개변수가 포함되어 있어 이를 변화시킬 수 있습니다. 이 양자 Circuit 집합은 바닥 상태를 근사하는 다양한 양자 상태를 준비할 수 있습니다.
- Estimator: 현재 변분 양자 상태에서 연산자 의 기댓값을 추정하는 수단입니다. 때로는 비용 함수라고 불리는 이 기댓값 자체가 목표인 경우도 있습니다. 또한 하나 이상의 기댓값으로부터 시작하여 더 복잡한 함수를 구하는 것이 목표인 경우도 있습니다.
- 고전 최적화기: 비용 함수를 최소화하기 위해 매개변수를 변화시키는 알고리즘입니다.
각 구성 요소를 더 자세히 살펴보겠습니다.
1.1 연산자 (해밀토니안)
VQE 문제의 핵심에는 관심 있는 시스템을 기술하는 연산자가 있습니다. 여기서는 이 연산자의 최솟값 고유값과 해당 고유벡터가 어떤 과학적 또는 비즈니스적 목적에 유용하다고 가정합니다. 예를 들어, 분자를 기술하는 화학 해밀토니안에서 연산자의 최솟값 고유값이 분자의 바닥 상태 에너지에 해당하고, 해당 고유 상태가 분자의 기하 구조 또는 전자 배치를 나타내는 경우가 있습니다. 또는 최적화할 특정 프로세스의 비용을 기술하는 연산자에서 고유 상태가 경로나 관행에 해당하는 경우도 있습니다. 물리학 같은 분야에서 "해밀토니안"은 거의 항상 물리 시스템의 에너지를 기술하는 연산자를 의미합니다. 하지만 양자 컴퓨팅에서는 비즈니스나 물류 문제를 기술하는 양자 연산자도 "해밀토니안"이라고 부르는 것이 일반적입니다. 여기서도 이 관례를 따릅니다.

물리적 또는 최적화 문제를 Qubit에 매핑하는 것은 일반적으로 간단하지 않지만, 그 세부 사항은 이 강좌의 초점이 아닙니다. 문제를 양자 연산자에 매핑하는 방법에 대한 일반적인 논의는 실전 양자 컴퓨팅에서 찾을 수 있습니다. 화학 문제를 양자 연산자로 매핑하는 방법에 대한 더 자세한 내용은 VQE를 이용한 양자 화학에서 확인할 수 있습니다.
이 강좌에서는 해밀토니안의 형태가 알려져 있다고 가정합니다. 예를 들어, 간단한 수소 분자의 해밀토니안(특정 활성 공간 가정과 Jordan-Wigner 매퍼를 사용하는 경우)은 다음과 같습니다.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-ibm-runtime scipy
from qiskit.quantum_info import SparsePauliOp
hamiltonian = SparsePauliOp(
[
"IIII",
"IIIZ",
"IZII",
"IIZI",
"ZIII",
"IZIZ",
"IIZZ",
"ZIIZ",
"IZZI",
"ZZII",
"ZIZI",
"YYYY",
"XXYY",
"YYXX",
"XXXX",
],
coeffs=[
-0.09820182 + 0.0j,
-0.1740751 + 0.0j,
-0.1740751 + 0.0j,
0.2242933 + 0.0j,
0.2242933 + 0.0j,
0.16891402 + 0.0j,
0.1210099 + 0.0j,
0.16631441 + 0.0j,
0.16631441 + 0.0j,
0.1210099 + 0.0j,
0.17504456 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
],
)
위 해밀토니안에는 서로 교환하지 않는 ZZII와 YYYY 같은 항들이 있습니다. 즉, ZZII를 평가하려면 3번 Qubit에서 Pauli Z 연산자를 측정해야 합니다. 하지만 YYYY를 평가하려면 같은 3번 Qubit에서 Pauli Y 연산자를 측정해야 합니다. 동일한 Qubit의 Y와 Z 연산자 사이에는 불확정성 관계가 있어 두 연산자를 동시에 측정할 수 없습니다. 이 점은 아래에서, 그리고 강좌 전반에 걸쳐 다시 다룰 것입니다.
위 해밀토니안은 행렬 연산자입니다. 연산자를 대각화하여 최저 에너지 고유값을 구하는 것은 어렵지 않습니다.
import numpy as np
A = np.array(hamiltonian)
eigenvalues, eigenvectors = np.linalg.eigh(A)
print("The ground state energy is ", min(eigenvalues), "hartrees")
The ground state energy is -1.1459778447627311 hartrees
브루트 포스 방식의 고전 고유값 분해기는 의약품이나 단백질처럼 매우 큰 원자 시스템의 에너지나 기하 구조를 기술하는 데 확장하기 어렵습니다. VQE는 이 문제에 양자 컴퓨팅을 활용하려는 초기 시도 중 하나입니다.
이 단원에서는 위보다 훨씬 큰 해밀토니안도 다루게 됩니다. 하지만 이 강좌의 후반부에서 VQE를 보완하거나 대체할 수 있는 더 발전된 도구들을 소개하기 전에, VQE의 한계를 먼저 시험하는 것은 비효율적일 것입니다.
1.2 Ansatz
"ansatz"는 독일어로 "접근법"을 의미합니다. 독일어에서 올바른 복수형은 "ansätze"이며, "ansatzes" 또는 "ansatze"로 쓰기도 합니다. VQE의 맥락에서 ansatz는 연구 중인 시스템의 바닥 상태를 가장 잘 근사하는 다중 qubit 파동 함수를 생성하는 데 사용하는 양자 Circuit으로, 연산자의 가장 낮은 기댓값을 만들어냅니다. 이 양자 Circuit에는 변분 매개변수(흔히 변수 벡터 로 묶음)가 포함됩니다.

변분 매개변수의 초깃값 을 선택합니다. ansatz의 단일 연산을 라고 부르겠습니다. IBM® 양자 컴퓨터의 모든 Qubit은 기본적으로 상태로 초기화됩니다. Circuit을 실행하면 Qubit의 상태는 다음과 같이 됩니다.
물리 시스템의 언어로 표현하면, 단순히 가장 낮은 에너지만 필요하다면 에너지를 여러 번 측정하여 최솟값을 취하면 됩니다. 하지만 일반적으로는 그 최저 에너지 또는 고유값을 생성하는 배치도 알고 싶습니다. 따라서 다음 단계는 양자 측정을 통해 해밀토니안의 기댓값을 추정하는 것입니다. 이 과정에는 많은 것이 포함됩니다. 하지만 에너지 를 측정할 확률 가 기댓값과 관련이 있다는 점에서 이 과정을 정성적으로 이해할 수 있습니다.
확률 는 고유 상태 와 현재 시스템 상태 간의 겹침과도 관련이 있습니다.
따라서 해밀토니안을 구성하는 Pauli 연산자들을 여러 번 측정함으로써 현재 시스템 상태 에서 해밀토니안의 기댓값을 추정할 수 있습니다. 다음 단계는 매개변수 를 변화시켜 시스템의 최저 에너지(바닥) 상태에 더 가까이 접근하는 것입니다. ansatz에 변분 매개변수가 포함되어 있기 때문에, 때로는 __변분 형식__이라고 부르기도 합니다.
변분 과정으로 넘어가기 전에, "좋은 초기 추측" 상태에서 시작하는 것이 유용한 경우가 많다는 점을 짚어두겠습니다. 시스템에 대해 충분히 알고 있다면 보다 더 나은 초기 추측을 할 수 있습니다. 예를 들어, 화학 응용에서는 Qubit을 Hartree-Fock 상태로 초기화하는 것이 일반적입니다. 변분 매개변수를 포함하지 않는 이 초기 추측을 __참조 상태__라고 합니다. 참조 상태를 생성하는 양자 Circuit을 라고 하겠습니다. 참조 상태와 나머지 ansatz를 구별해야 할 때는 다음을 사용합니다: 동등하게 표현하면
1.3 Estimator
특정 변분 상태 에서 해밀토니안의 기댓값을 추정하는 방법이 필요합니다. 연산자 전체를 직접 측정할 수 있다면, 번 측정하여 평균을 구하는 것만큼 간단할 것입니다.
여기서 기호는 극한에서만 이 기댓값이 정확하다는 것을 상기시켜 줍니다. 하지만 Circuit에서 수천 번의 측정을 수행하면 기댓값의 샘플링 오차는 꽤 낮아집니다. 매우 정밀한 계산에서는 노이즈 같은 다른 고려 사항도 문제가 됩니다.
하지만 일반적으로 를 한 번에 측정하는 것은 불가능합니다. 에는 서로 교환하지 않는 Pauli X, Y, Z 연산자가 여러 개 포함될 수 있습니다. 따라서 해밀토니안을 동시에 측정할 수 있는 연산자 그룹으로 분해해야 하며, 각 그룹을 별도로 추정한 후 결과를 합산하여 기댓값을 구해야 합니다. 이에 대해서는 다음 단원에서 고전적 접근과 양자적 접근의 확장성을 논의할 때 더 자세히 다룰 것입니다. 이처럼 측정의 복잡성 때문에 추정 수행에 효율적인 코드가 필요합니다. 이 단원 이후로는 이 목적을 위해 Qiskit Runtime 기본 요소인 Estimator를 사용할 것입니다.
1.4 고전 최적화기
고전 최적화기는 목적 함수(일반적으로 최솟값)의 극값을 찾기 위한 고전 알고리즘입니다. 관심 있는 함수를 최소화하는 매개변수 집합을 찾기 위해 가능한 매개변수 공간을 탐색합니다. 크게 기울기 정보를 활용하는 기울기 기반 방법과 블랙박스 최적화기로 작동하는 기울기 없는 방법으로 분류할 수 있습니다. 고전 최적화기의 선택은 특히 양자 하드웨어의 노이즈가 있는 환경에서 알고리즘 성능에 크게 영향을 미칠 수 있습니다. 이 분야에서 널리 사용되는 최적화기로는 노이즈 환경에서 유망한 결과를 보여준 Adam, AMSGrad, SPSA가 있습니다. 더 전통적인 최적화기로는 COBYLA와 SLSQP가 있습니다.
일반적인 워크플로우(섹션 3.3에서 시연)는 scipy의 minimize 함수와 같은 최소화 함수 내에서 이러한 알고리즘 중 하나를 방법으로 사용하는 것입니다. 이 함수는 다음을 인수로 받습니다.
- 최소화할 함수. 흔히 에너지 기댓값이 사용됩니다. 하지만 일반적으로는 "비용 함수"라고 부릅니다.
- 탐색을 시작할 매개변수 집합. 흔히 또는 라고 합니다.
- 비용 함수의 인수를 포함한 인수들. Qiskit을 이용한 양자 컴퓨팅에서는 ansatz, 해밀토니안, 다음 소절에서 더 자세히 다루는 Estimator 프리미티브가 포함됩니다.
- 최소화 '방법'. 매개변수 공간을 탐색하는 데 사용되는 특정 알고리즘을 의미합니다. 여기서 예를 들어 COBYLA 또는 SLSQP를 지정합니다.
- 옵션. 사용 가능한 옵션은 방법에 따라 다를 수 있습니다. 하지만 거의 모든 방법에서 공통적으로 포함되는 예는 탐색 종료 전 최적화기의 최대 반복 횟수인 'maxiter'입니다.

각 반복 단계에서 해밀토니안의 기댓값은 많은 측정을 통해 추정됩니다. 이 추정 에너지는 비용 함수가 반환하고, 최소화기는 에너지 지형에 대한 정보를 업데이트합니다. 다음 단계를 선택하기 위해 최적화기가 수행하는 작업은 방법마다 다릅니다. 일부는 기울기를 사용하여 가장 급격한 하강 방향을 선택합니다. 다른 것들은 노이즈를 고려하여 그 방향으로 실제 에너지가 감소한다고 받아들이기 전에 비용이 큰 폭으로 감소하도록 요구할 수도 있습니다.
# Example syntax for minimization
# from scipy.optimize import minimize
# res = minimize(cost_func, x0, args=(ansatz, hamiltonian, estimator), method="cobyla",
# options={'maxiter': 200})
1.5 변분 원리
이 맥락에서 변분 원리는 매우 중요합니다. 변분 원리는 어떤 변분 파동 함수도 바닥 상태 파동 함수가 만들어내는 에너지(또는 비용) 기댓값보다 낮은 값을 생성할 수 없다고 말합니다. 수학적으로,
의 모든 고유 상태 집합 이 힐베르트 공간의 완전 기저를 이룬다는 점에 주목하면 이를 쉽게 검증할 수 있습니다. 다시 말해, 임의의 상태, 특히 는 의 이 고유 상태들의 가중(정규화된) 합으로 쓸 수 있습니다.
여기서 는 결정될 상수이고, 입니다. 이를 독자에게 연습 문제로 남깁니다. 하지만 그 함의에 주목하세요: 가장 낮은 에너지 기댓값을 만들어내는 변분 상태가 바로 실제 바닥 상태의 최선의 추정값입니다.
이해도 확인
임의의 변분 상태 에 대해 임을 수학적으로 증명하세요.
Answer
에너지 고유 상태로 변분 상태를 전개한 표현을 이용하면,
변분 에너지 기댓값을 다음과 같이 쓸 수 있습니다.
모든 계수에 대해 이 성립합니다. 따라서 다음을 쓸 수 있습니다.
2. 고전적 워크플로와의 비교
N행 N열의 행렬이 있다고 가정해 봅시다. 이 행렬이 너무 커서 정확한 대각화가 불가능하다고 가정합시다. 또한 문제에 대해 충분히 알고 있어서 목표 고유벡터의 전반적인 구조를 어느 정도 추측할 수 있고, 초기 추측값과 유사한 상태들을 탐색하여 비용/에너지를 더 낮출 수 있는지 확인하고 싶다고 가정합시다. 이것이 변분적 접근 방식이며, 정확한 대각화가 불가능할 때 사용되는 방법 중 하나입니다.
2.1 고전적 워크플로
고전 컴퓨터를 사용하면 다음과 같이 진행됩니다:
- 변분할 파라미터 를 가진 추측 상태 를 만듭니다. 이 초기 추측값이 무작위일 수도 있지만, 그것은 권장되지 않습니다. 우리는 해당 문제에 대한 지식을 활용하여 추측값을 최대한 맞춤화하고 싶습니다.
- 해당 상태에서 연산자의 기댓값을 계산합니다:
- 변분 파라미터를 변경하고 반복합니다: .
- 변분 부분공간에서 가능한 상태들의 에너지 지형에 대해 축적된 정보를 활용하여 점점 더 나은 추측값을 만들고 목표 상태에 접근합니다. 변분 원리는 우리의 변분 상태가 목표 바닥 상태의 고유값보다 낮은 고유값을 산출할 수 없음을 보장합니다. 따라서 기댓값이 낮을수록 바닥 상태에 대한 근사가 더 좋습니다:
이 접근 방식에서 각 단계의 어려움을 살펴봅시다. 파라미터를 설정하거나 업데이트하는 것은 계산적으로 쉽습니다. 어려움은 유용하고 물리적으로 동기가 부여된 초기 파라미터를 선택하는 데 있습니다. 이전 반복에서 축적된 정보를 이용하여 바닥 상태에 가까워지도록 파라미터를 업데이트하는 것은 자명하지 않습니다. 그러나 이를 꽤 효율적으로 수행하는 고전 최적화 알고리즘이 존재합니다. 이 고전 최적화는 많은 반복이 필요할 수 있기 때문에 비용이 많이 들 수 있으며, 최악의 경우 반복 횟수가 N에 대해 지수적으로 증가할 수 있습니다. 계산적으로 가장 비용이 높은 단계는 주어진 상태 를 사용하여 행렬의 기댓값을 계산하는 것입니다:
행렬은 -원소 벡터에 작용해야 하며, 이는 최악의 경우 번의 곱셈 연산에 해당합니다. 이 작업은 파라미터의 각 반복마다 수행되어야 합니다. 매우 큰 행렬의 경우, 이는 높은 계산 비용을 수반합니다.
2.2 양자 워크플로와 교환 가능한 파울리 그룹
이제 계산의 이 부분을 양자 컴퓨터에 맡긴다고 상상해 보세요. 이 기댓값을 직접 계산하는 대신, 변분 ansatz를 사용하여 양자 컴퓨터에 상태 를 준비하고 측정을 수행하여 기댓값을 추정합니다.
그것이 들리는 것만큼 쉽지는 않습니다. 는 일반적으로 측정하기 쉽지 않습니다. 예를 들어 서로 교환 불가능한 파울리 X, Y, Z 연산자들로 구성될 수 있습니다. 그러나 는 각각 측정하기 쉬운 항들 의 선형 조합으로 쓸 수 있습니다 (예: 파울리 연산자 또는 큐비트별 교환 가능한 파울리 연산자 그룹). 어떤 상태 에 대한 의 기댓값은 구성 항들 의 기댓값의 가중합입니다. 이 표현식은 임의의 상태 에 대해 성립하지만, 우리는 특히 변분 상태 에 이를 적용할 것입니다.
여기서 는 IZZX…XIYX와 같은 파울리 문자열이거나 서로 교환 가능한 여러 그런 문자열입니다. 따라서 양자 컴퓨터에서의 측정 실제에 더 가깝게 기댓값을 표현하면 다음과 같습니다:
그리고 변분 파동 함수의 맥락에서:
각 항 는 번 측정되어 측정 샘플 ()를 산출하고, 기댓값 와 표준 편차 를 반환합니다. 이 항들을 합산하고 합을 통해 오차를 전파하여 전체 기댓값 와 표준 편차 를 얻을 수 있습니다.
이 방법은 대규모 곱셈이나 에 비례하는 과정을 필요로 하지 않습니다. 대신 양자 컴퓨터에서 여러 번의 측정이 필요합니다. 그 측정 횟수가 너무 많지 않다면, 이 접근 방식은 효율적일 수 있습니다. 이것이 VQE의 양자 부분입니다.
하지만 이것이 왜 효율적이지 않을 수 있는지에 대해서도 이야기해 봅시다. 많은 측정이 필요한 한 가지 이유는 매우 높은 정밀도의 계산을 위해 추정값의 통계적 불확실성을 줄이기 위해서입니다. 또 다른 이유는 전체 행렬을 표현하는 데 필요한 파울리 문자열의 수입니다. 파울리 행렬(항등 포함: X, Y, Z, I)이 주어진 차원의 모든 연산자 공간을 생성하므로, 앞서 했던 것처럼 우리의 행렬을 파울리 연산자의 가중합으로 쓸 수 있음이 보장됩니다.
여기서 는 IZZX…XIYX와 같이 시스템을 나타내는 모든 큐비트에 작용하는 파울리 문자열이거나, 서로 교환 가능한 여러 그런 문자열입니다. Qiskit은 리틀 엔디언 표기법을 사용하며, 오른쪽에서 번째 파울리 연산자가 번째 Qubit에 작용합니다. 따라서 일련의 파울리 연산자를 측정하여 연산자를 측정할 수 있습니다.
그러나 그 파울리 연산자들을 모두 동시에 측정할 수는 없습니다. (항등을 제외한) 파울리 연산자는 동일한 Qubit와 연관된 경우 서로 교환되지 않습니다. 예를 들어 IZIZ와 ZZXZ는 동시에 측정할 수 있는데, 세 번째 Qubit에 대해 I와 Z를 동시에 측정할 수 있고, 첫 번째 Qubit에 대해 I와 X를 동시에 알 수 있기 때문입니다. 그러나 ZZZZ와 ZZZX는 동시에 측정할 수 없습니다. Z와 X가 교환되지 않으며 둘 다 0번째 Qubit에 작용하기 때문입니다. 경험이 많은 독자라면 두 파울리 연산자 그룹이 집합으로서는 교환 가능할 수 있지만, 각 개별 Qubit의 측정은 교환 불가능할 수도 있다는 점을 떠올릴 수 있을 것입니다. Estimator는 Qubit별로 교환 가능한 연산자를 그룹화하는 방식에 해당하는 텐서곱 파울리 측정(기저 회전을 통해)을 가정합니다. 따라서 Estimator를 사용하여 두 개의 파울리 연산자 문자열(A와 B)을 동시에 추정하려면, A와 B의 각 Qubit에 대한 파울리 연산자들이 교환 가능해야 합니다. 이는 ZZZZ와 ZZXX도 동시에 측정할 수 없음을 의미합니다.

따라서 행렬 를 서로 다른 Qubit에 작용하는 파울리 연산자들의 합으로 분해합니다. 그 합의 일부 원소들은 한꺼번에 측정될 수 있으며, 이를 교환 가능한 파울리 그룹이라고 합니다. 교환 불가능한 항의 수에 따라 많은 그룹이 필요할 수 있습니다. 교환 가능한 파울리 문자열 그룹의 수를 라고 합시다. 가 작으면 이 방법이 잘 작동할 수 있습니다. 에 수백만 개의 그룹이 있다면, 이 방법은 유용하지 않을 것입니다.
기댓값 추정에 필요한 과정들은 Estimator라는 Qiskit Runtime 프리미티브에 통합되어 있습니다. Estimator에 대한 자세한 내용은 IBM Quantum® Documentation의 API 참조를 참고하세요. Estimator를 직접 사용할 수 있지만, Estimator는 가장 낮은 에너지 고유값 이상의 정보를 반환합니다. 예를 들어, 앙상블 표준 오차에 대한 정보도 반환합니다. 따라서 최소화 문제의 맥락에서, Estimator가 비용 함수 안에 포함된 경우를 자주 볼 수 있습니다. Estimator의 입력 및 출력에 대한 자세한 내용은 IBM Quantum Documentation의 이 가이드를 참고하세요.
상태에 사용된 파라미터 집합 에 대한 기댓값(또는 비용 함수)을 기록하고, 파라미터를 업데이트합니다. 시간이 지남에 따라, 추정한 기댓값이나 비용 함수 값을 활용하여 ansatz가 샘플링하는 상태의 부분공간에서 비용 함수의 기울기를 근사할 수 있습니다. 기울기 기반 최적화기와 기울기를 사용하지 않는 최적화기 모두 존재합니다. 두 방법 모두 다수의 국소 최솟값이나 *황무지 고원(barren plateau)*이라 불리는 기울기가 거의 0에 가까운 광대한 파라미터 공간 영역과 같은 학습 가능성 문제를 겪을 수 있습니다.

2.3 계산 비용을 결정하는 요인
VQE가 모든 어려운 양자 화학 문제를 해결해 주지는 않습니다. 그렇지 않습니다. 하지만 모든 계산에서 더 나은 성능을 내는 것이 목적이 아닙니다. 계산 비용을 결정하는 요소가 달라졌습니다.

우리는 복잡도가 행렬 차원에만 의존하는 과정에서, 필요한 정밀도와 행렬을 구성하는 교환 불가능한 파울리 연산자의 수에 의존하는 과정으로 전환하였습니다. 마지막 부분은 고전 컴퓨팅에 유사한 개념이 없습니다.
이러한 의존성을 바탕으로, 희소 행렬이나 교환 불가능한 파울리 문자열이 적은 행렬의 경우 이 과정이 유용할 수 있습니다. 상호작용하는 스핀 시스템이 이에 해당합니다. 밀집 행렬의 경우에는 덜 유용할 수 있습니다. 예를 들어, 화학 시스템은 종종 수백, 수천, 심지어 수백만 개의 파울리 문자열을 포함하는 Hamiltonian을 가진다는 것이 알려져 있습니다. 이 항들의 수를 줄이기 위한 흥미로운 연구들이 진행되어 왔습니다. 하지만 화학 시스템은 이 과정에서 나중에 다룰 다른 알고리즘들에 더 적합할 수도 있습니다.
이해도 점검
네 개의 Qubit에 대한 Hamiltonian이 다음 항들을 포함한다고 가정합니다:
IIXX, IIXZ, IIZZ, IZXZ, IXXZ, ZZXZ, XZXZ, ZIXZ, ZZZZ, XXXX
모든 항이 그룹 내에서 동시에 측정될 수 있도록 이 항들을 그룹으로 분류하고 싶습니다. 모든 항을 포함하는 최소한의 그룹 수는 얼마입니까?
Answer
4개의 그룹으로 가능합니다. 이러한 해는 일반적으로 유일하지 않습니다.
IIXX, XXXX, IIZZ, ZZZZ
IIXZ, IZXZ, ZIXZ, ZZXZ
IXXZ
XZXZ
VQE를 이용한 양자 화학에서 일반적으로 어떤 것이 더 어렵게 만드나요: Hamiltonian의 항 수 아니면 좋은 ansatz 찾기?
Answer
화학적 맥락에 고도로 최적화된 ansätze가 존재합니다. Hamiltonian의 항 수, 즉 필요한 측정 횟수가 일반적으로 더 많은 문제를 일으킵니다.
3. 예시 해밀토니안
이 알고리즘을 작은 해밀토니안 행렬로 직접 실습해 보겠습니다. 각 단계에서 무슨 일이 일어나는지 확인할 수 있도록, Qiskit 패턴 프레임워크를 사용합니다.
-1단계: 문제를 양자 Circuit과 연산자로 매핑 -2단계: 대상 하드웨어에 맞게 최적화 -3단계: 대상 하드웨어에서 실행 -4단계: 결과 후처리
3.1 1단계: 문제를 양자 Circuit과 연산자로 매핑
위의 화학 컨텍스트에서 정의한 것을 사용하겠습니다. 먼저 일반 임포트부터 시작합니다.
# General imports
import numpy as np
# SciPy minimizer routine
from scipy.optimize import minimize
# Plotting functions
import matplotlib.pyplot as plt
다시 한번, 관심 있는 해밀토니안은 이미 알려져 있다고 가정합니다. 이 코스에서 나중에 다룰 다른 방법들이 더 큰 문제를 더 효율적으로 풀 수 있기 때문에, 여기서는 매우 작은 해밀토니안을 사용합니다.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
hamiltonian = SparsePauliOp.from_list(
[("YZ", 0.3980), ("ZI", -0.3980), ("ZZ", -0.0113), ("XX", 0.1810)]
)
A = np.array(hamiltonian)
eigenvalues, eigenvectors = np.linalg.eigh(A)
print("The ground state energy is ", min(eigenvalues))
The ground state energy is -0.702930394459531
Qiskit에는 미리 만들어진 ansatz 선택지가 많이 있습니다. 여기서는 efficient_su2를 사용하겠습니다.
# Pre-defined ansatz circuit and operator class for Hamiltonian
from qiskit.circuit.library import efficient_su2
# Note that it is more common to place initial 'h' gates outside the ansatz.
# Here we specifically wanted this layer structure.
ansatz = efficient_su2(
hamiltonian.num_qubits, su2_gates=["h", "rz", "y"], entanglement="circular", reps=1
)
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.decompose().draw("mpl", style="iqp")
This circuit has 4 parameters
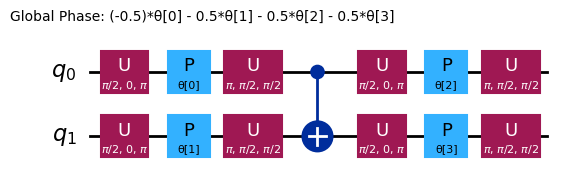
ansätze마다 서로 다른 얽힘 구조와 회전 Gate를 가집니다. 여기서 보이는 ansatz는 얽힘을 위해 CNOT Gate를 사용하고, 회전을 위해 Y Gate와 매개변수화된 RZ Gate를 모두 사용합니다. 이 매개변수 공간의 크기에 주목하세요. 즉, 비용 함수를 4개의 변수(RZ Gate의 매개변수)에 대해 최소화해야 합니다. 이를 더 확장할 수는 있지만 무한정 확장할 수는 없습니다. 예를 들어 4 qubit 문제에서 efficient_su2의 기본값인 3 reps를 사용하면 16개의 변분 매개변수가 생성됩니다.
3.2 2단계: 대상 하드웨어에 맞게 최적화
ansatz는 친숙한 Gate를 사용하여 작성되었지만, 각 양자 컴퓨터에서 구현 가능한 기본 Gate를 활용하려면 Circuit을 Transpile해야 합니다. 가장 바쁘지 않은 Backend를 선택합니다.
# runtime imports
from qiskit_ibm_runtime import QiskitRuntimeService, Session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# To run on hardware, select the backend with the fewest number of jobs in the queue
service = QiskitRuntimeService()
backend = service.least_busy(operational=True, simulator=False)
print(backend)
<IBMBackend('ibm_torino')>
이제 이 하드웨어에 맞게 Circuit을 Transpile하고 변환된 ansatz를 시각화할 수 있습니다.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")

사용된 Gate가 변경되었고, 추상 Circuit의 Qubit들이 양자 컴퓨터의 서로 다른 번호의 Qubit에 매핑되었음을 알 수 있습니다. 결과가 의미 있으려면 해밀토니안도 동일하게 매핑해야 합니다.
hamiltonian_isa = hamiltonian.apply_layout(layout=ansatz_isa.layout)
3.3 3단계: 대상 하드웨어에서 실행
3.3.1 값 출력
여기서는 이전 단계에서 구성한 구조들, 즉 매개변수, ansatz, 해밀토니안을 인수로 받는 비용 함수를 정의합니다. 아직 정의하지 않은 Estimator도 사용합니다. 수렴 동작을 확인할 수 있도록 비용 함수의 이력을 추적하는 코드도 포함합니다.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from Estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
문제에 대한 지식과 목표 상태의 특성을 바탕으로 초기 매개변수 값을 선택할 수 있다면 매우 유리합니다. 여기서는 그러한 지식이 없다고 가정하고 무작위 초기값을 사용합니다.
x0 = 2 * np.pi * np.random.random(num_params)
# This required 13 min, 20 s QPU time on an Eagle processor, 28 min total time.
with Session(backend=backend) as session:
estimator = Estimator(mode=session)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 50},
)
Iters. done: 1 [Current cost: 0.010575798722044727]
Iters. done: 2 [Current cost: 0.004040015974440895]
Iters. done: 3 [Current cost: 0.0020213258785942503]
Iters. done: 4 [Current cost: 0.18723082446726014]
Iters. done: 5 [Current cost: -0.2746792152068885]
Iters. done: 6 [Current cost: -0.3094547651648519]
Iters. done: 7 [Current cost: -0.05281985428356641]
Iters. done: 8 [Current cost: 0.00808560303514377]
Iters. done: 9 [Current cost: -0.0014821685303514388]
Iters. done: 10 [Current cost: -0.004759824281150161]
Iters. done: 11 [Current cost: 0.09942328705995292]
Iters. done: 12 [Current cost: 0.01092366214057508]
Iters. done: 13 [Current cost: 0.05017497496069776]
Iters. done: 14 [Current cost: 0.13028868414310696]
Iters. done: 15 [Current cost: 0.013747803514376994]
Iters. done: 16 [Current cost: 0.2583072432944498]
Iters. done: 17 [Current cost: -0.14422125655131562]
Iters. done: 18 [Current cost: -0.0004950150347678081]
Iters. done: 19 [Current cost: 0.00681082268370607]
Iters. done: 20 [Current cost: -0.0023377795527156544]
Iters. done: 21 [Current cost: 0.6027665591169237]
Iters. done: 22 [Current cost: 0.00596641373801917]
Iters. done: 23 [Current cost: -0.008318769968051117]
Iters. done: 24 [Current cost: -0.00026683306709265246]
Iters. done: 25 [Current cost: -0.007648222843450479]
Iters. done: 26 [Current cost: 0.004121086261980831]
Iters. done: 27 [Current cost: -0.004075019968051117]
Iters. done: 28 [Current cost: -0.004419369009584665]
Iters. done: 29 [Current cost: 0.213185460054037]
Iters. done: 30 [Current cost: -0.06505919572162797]
Iters. done: 31 [Current cost: -0.5334241316590271]
Iters. done: 32 [Current cost: 0.00218370607028754]
Iters. done: 33 [Current cost: 0.09579352143666908]
Iters. done: 34 [Current cost: -0.009274800319488819]
Iters. done: 35 [Current cost: -0.44395141360688106]
Iters. done: 36 [Current cost: 0.011747104632587858]
Iters. done: 37 [Current cost: -0.003344149361022364]
Iters. done: 38 [Current cost: 0.19138183916486304]
Iters. done: 39 [Current cost: 0.013513931813145209]
원시 출력값을 확인할 수 있습니다.
res
message: Return from COBYLA because the trust region radius reaches its lower bound.
success: True
status: 0
fun: -0.5334241316590271
x: [ 1.024e+00 6.459e+00 3.625e+00 4.007e+00]
nfev: 39
maxcv: 0.0
3.4 Step 4: Post-process results
절차가 올바르게 종료되었다면, 딕셔너리의 값은 각각 해의 벡터와 총 함수 평가 횟수와 일치해야 합니다. 이를 쉽게 확인할 수 있습니다.
cost_history_dict
{'prev_vector': array([1.02397956, 6.45886604, 3.62479262, 4.00744128]),
'iters': 39,
'cost_history': [np.float64(0.010575798722044727),
np.float64(0.004040015974440895),
np.float64(0.0020213258785942503),
np.float64(0.18723082446726014),
np.float64(-0.2746792152068885),
np.float64(-0.3094547651648519),
np.float64(-0.05281985428356641),
np.float64(0.00808560303514377),
np.float64(-0.0014821685303514388),
np.float64(-0.004759824281150161),
np.float64(0.09942328705995292),
np.float64(0.01092366214057508),
np.float64(0.05017497496069776),
np.float64(0.13028868414310696),
np.float64(0.013747803514376994),
np.float64(0.2583072432944498),
np.float64(-0.14422125655131562),
np.float64(-0.0004950150347678081),
np.float64(0.00681082268370607),
np.float64(-0.0023377795527156544),
np.float64(0.6027665591169237),
np.float64(0.00596641373801917),
np.float64(-0.008318769968051117),
np.float64(-0.00026683306709265246),
np.float64(-0.007648222843450479),
np.float64(0.004121086261980831),
np.float64(-0.004075019968051117),
np.float64(-0.004419369009584665),
np.float64(0.213185460054037),
np.float64(-0.06505919572162797),
np.float64(-0.5334241316590271),
np.float64(0.00218370607028754),
np.float64(0.09579352143666908),
np.float64(-0.009274800319488819),
np.float64(-0.44395141360688106),
np.float64(0.011747104632587858),
np.float64(-0.003344149361022364),
np.float64(0.19138183916486304),
np.float64(0.013513931813145209)]}
fig, ax = plt.subplots()
x = np.linspace(0, 10, 50)
# Define the constant function
constant = -0.7029
y_constant = np.full_like(x, constant)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

IBM Quantum에는 VQE와 관련된 다른 역량 강화 콘텐츠도 있습니다. VQE를 실제로 적용해 보고 싶다면 다음 튜토리얼을 참고하세요: Ground-state energy estimation of the Heisenberg chain with VQE. 분자 Hamiltonian 생성에 대해 더 알고 싶다면 Quantum chemistry with VQE 과정의 해당 레슨을 참고하세요. VQE와 같은 변분 알고리즘의 동작 원리를 더 깊이 이해하고 싶다면 Variational Algorithm Design 과정을 추천합니다.
Check your understanding
이 섹션에서는 Hamiltonian로부터 바닥 상태 에너지를 계산했습니다. 이를 분자의 기하 구조를 결정하는 데 적용하려면 어떻게 확장해야 할까요?
Answer
원자 간 거리와 결합 각도에 대한 변수를 도입하고 이를 변분 파라미터로 활용해야 합니다. 이 변수들을 변화시킬 때마다 새로운 Hamiltonian이 생성됩니다(에너지를 기술하는 연산자는 기하 구조에 따라 달라지기 때문입니다). 각 Hamiltonian을 Qubit에 매핑한 뒤, 위에서 수행한 것과 같은 최적화를 반복해야 합니다. 수렴된 모든 최적화 문제 중에서 가장 낮은 에너지를 산출한 기하 구조가 자연이 선택하는 구조가 됩니다. 이는 위에서 다룬 것보다 훨씬 복잡한 과정입니다. 가장 단순한 분자인 에 대한 이러한 계산은 여기에서 확인할 수 있습니다.
4. VQE와 다른 방법들의 관계
이 섹션에서는 원래의 VQE 방식이 가진 장단점을 검토하고, 보다 최근에 등장한 알고리즘들과의 관계를 살펴봅니다.
4.1 VQE의 강점과 약점
강점 중 일부는 이미 언급했습니다. 여기에는 다음이 포함됩니다.
- 현대 하드웨어에 대한 적합성: 일부 양자 알고리즘은 훨씬 낮은 오류율, 즉 대규모 내결함성에 가까운 수준을 요구합니다. VQE는 그렇지 않으며, 현재의 양자 컴퓨터에서 구현할 수 있습니다.
- 얕은 Circuit: VQE는 비교적 얕은 양자 Circuit을 사용하는 경우가 많습니다. 이로 인해 VQE는 누적된 Gate 오류에 덜 취약하며, 많은 오류 완화 기법에 적합합니다. 물론 Circuit이 항상 얕은 것은 아니며, 이는 사용하는 ansatz에 따라 달라집니다.
- 다용도성: VQE는 (원칙적으로) 고유값/고유벡터 문제로 표현할 수 있는 모든 문제에 적용할 수 있습니다. 다만 VQE를 비실용적이거나 불리하게 만드는 여러 제약이 있습니다. 이 중 일부는 아래에서 다시 정리합니다.
VQE의 약점과 실용적이지 않은 경우들도 위에서 일부 설명했습니다. 여기에는 다음이 포함됩니다.
- 휴리스틱 특성: VQE는 정확한 바닥 상태 에너지로의 수렴을 보장하지 않으며, 그 성능은 ansatz와 최적화 방법의 선택에 따라 달라집니다[1-2]. 목표 바닥 상태에 필요한 얽힘을 갖추지 못한 부적절한 ansatz를 선택하면, 어떤 고전 최적화 알고리즘도 해당 바닥 상태에 도달할 수 없습니다.
- 잠재적으로 많은 파라미터: 매우 표현력이 높은 ansatz는 파라미터 수가 너무 많아 최소화 반복에 상당한 시간이 소요될 수 있습니다.
- 높은 측정 오버헤드: VQE에서는 Estimator를 사용하여 해밀토니안 각 항의 기댓값을 추정합니다. 관심 있는 대부분의 해밀토니안에는 동시에 추정할 수 없는 항들이 포함되어 있습니다. 이로 인해 복잡한 해밀토니안을 가진 대형 시스템에서 VQE는 자원 집약적인 방법이 될 수 있습니다[1].
- 잡음의 영향: 고전 최적화 알고리즘이 최솟값을 탐색할 때, 잡음이 있는 계산은 알고리즘을 혼란스럽게 하여 진정한 최솟값에서 멀어지게 하거나 수렴을 지연시킬 수 있습니다. 이에 대한 한 가지 해결책은 IBM의 최신 오류 완화 및 오류 억제 기법[2-3]을 활용하는 것입니다.
- 불모 고원(Barren plateau): 기울기가 소실되는 이 영역[2-3]은 잡음이 없는 경우에도 존재하지만, 잡음이 있으면 더욱 문제가 됩니다. 이 불모 영역에서는 잡음으로 인한 기댓값 변화가 파라미터 업데이트로 인한 변화보다 클 수 있기 때문입니다.
4.2 다른 접근법과의 관계
Adapt-VQE
ADAPT-VQE (Adaptive Derivative-Assembled Pseudo-Trotter Variational Quantum Eigensolver)는 원래 VQE 알고리즘을 개선한 방법으로, 특히 양자 화학 분야의 양자 시뮬레이션에서 효율성, 정확도, 확장성을 향상시키도록 설계되었습니다.
이 강의 전반에 걸쳐 설명한 원래의 VQE 알고리즘은 미리 정의된 고정 ansatz를 사용하여 시스템의 바닥 상태를 근사합니다. 우리는 1회 반복과 Y 및 RZ 회전 Gate를 사용하는 efficient_su2를 사용했습니다. RZ Gate의 파라미터는 변경되었지만, 이 ansatz의 구조와 사용된 Gate는 변경되지 않았습니다.
ADAPT-VQE는 적응형 ansatz 구성을 통해 VQE의 한계를 해결합니다. 고정된 ansatz로 시작하는 대신, ADAPT-VQE는 ansatz를 반복적으로 동적으로 구축합니다. 각 단계에서 미리 정의된 풀(예: 페르미온 여기 연산자)에서 에너지에 대한 기울기가 가장 큰 연산자를 선택합니다. 이를 통해 가장 영향력 있는 연산자만 추가되어 간결하고 효율적인 ansatz가 만들어집니다[4-6]. 이 접근법은 다음과 같은 여러 이점을 가져올 수 있습니다.
- Circuit 깊이 감소: ansatz를 점진적으로 확장하고 필요한 연산자에만 집중함으로써, ADAPT-VQE는 기존 VQE 방식에 비해 Gate 연산 수를 최소화합니다[5,7].
- 정확도 향상: 적응형 특성 덕분에 ADAPT-VQE는 각 단계에서 더 많은 상관 에너지를 회복할 수 있어, 기존 VQE가 어려움을 겪는 강하게 상관된 시스템에 특히 효과적입니다[8,9].
- 확장성 및 잡음 견고성: 간결한 ansatz는 Gate 오류의 누적을 줄이고, 계산 오버헤드를 낮추며, 최소화해야 할 변분 파라미터 수를 제한합니다.
ADAPT-VQE도 완벽하지는 않습니다. 경우에 따라 지역 최솟값에 갇히거나 느려질 수 있으며, 과잉 파라미터화 문제가 발생할 수도 있습니다. 또한 기울기 계산과 다양한 Gate 구조에 대한 파라미터 최적화가 필요하기 때문에, 상당한 자원이 소모될 수 있습니다.
양자 위상 추정(QPE)
QPE는 목적 면에서 VQE와 유사하지만, 구현 방식은 매우 다릅니다. QPE는 일반적으로 깊은 양자 Circuit과 높은 수준의 결맞음이 필요하기 때문에 내결함성 양자 컴퓨터를 요구합니다. QPE가 구현되면 VQE보다 더 정밀한 결과를 얻을 수 있습니다. 두 방법의 차이를 설명하는 한 가지 방식은 Circuit 깊이에 따른 정밀도입니다. QPE는 으로 확장되는 Circuit 깊이로 정밀도 을 달성합니다[10]. VQE는 동일한 정밀도를 달성하기 위해 회의 샘플링이 필요합니다[10,11].
이 강의에서 다루는 Krylov, SQD, QSCI 등
VQE는 양자 컴퓨터를 운용하는 데만 고전 컴퓨터를 사용하는 것이 아니라, 알고리즘의 실질적인 부분에도 고전 컴퓨터에 의존하는 양자 알고리즘을 확립하는 데 기여했습니다. 이 강의의 나머지 부분에서는 그러한 알고리즘 중 여럿을 집중적으로 다룹니다. 여기서는 VQE와 비교하기 위해 몇 가지를 간략히 소개합니다. 자세한 내용은 이후 강의에서 설명합니다.
Krylov 양자 대각화(KQD)
__Krylov 부분공간 방법__은 행렬을 부분공간에 투영하여 차원을 줄이고 다루기 쉽게 만드는 방법으로, 가장 중요한 특징은 유지합니다. 이 방법의 핵심 기법 중 하나는 이러한 특징을 보존하는 부분공간을 생성하는 것인데, 이 부분공간 생성은 양자 컴퓨터에서 잘 확립된 방법인 __Trotterization__과 밀접하게 관련되어 있습니다.
양자 Krylov 방법에는 몇 가지 변형이 있지만, 일반적인 접근 방식은 다음과 같습니다.
- Trotterization을 통해 양자 컴퓨터로 부분공간(Krylov 부분공간)을 생성합니다.
- 관심 행렬을 해당 Krylov 부분공간에 투영합니다.
- 투영된 새로운 해밀토니안을 고전 컴퓨터로 대각화합니다.
샘플링 기반 양자 대각화(SQD)
__샘플링 기반 양자 대각화(SQD)__는 대각화할 행렬의 차원을 줄이면서 핵심 특징을 보존하려 한다는 점에서 Krylov 방법과 관련이 있습니다. SQD는 다음과 같은 방식으로 이를 수행합니다.
- 바닥 상태에 대한 초기 추정치로 시작하여, 시스템을 해당 바닥 상태로 준비합니다.
- Sampler를 사용하여 이 상태를 구성하는 비트열을 샘플링합니다.
- Sampler에서 얻은 계산 기저 상태들의 집합을 관심 행렬을 투영할 부분공간으로 사용합니다.
- 고전 컴퓨터로 더 작게 투영된 행렬을 대각화합니다.
이 방법은 알고리즘의 실질적인 부분에서 고전 컴퓨팅과 양자 컴퓨팅을 모두 활용한다는 점에서 VQE와 관련이 있습니다. 두 방법 모두 좋은 초기 추정값 또는 ansatz를 준비해야 한다는 공통 요건도 있습니다. 하지만 SQD에서 고전 컴퓨터와 양자 컴퓨터 간의 작업 분배는 Krylov 방법과 더 유사합니다.
실제로 Krylov 방법과 SQD는 최근 샘플링 기반 Krylov 양자 대각화(SKQD) 방법[12]으로 결합되었습니다.
양자 부분공간 구성 상호작용
양자 선택 구성 상호작용(QSCI)[13]은 시도 파동함수를 샘플링하여 중요한 계산 기저 상태를 파악하고, 이를 고전 대각화를 위한 부분공간으로 구성함으로써 해밀토니안의 근사 바닥 상태를 생성하는 알고리즘입니다. SQD와 QSCI 모두 양자 컴퓨터를 사용하여 축소된 부분공간을 구성합니다. QSCI의 추가적인 강점은 상태 준비에 있으며, 특히 화학 문제의 맥락에서 두드러집니다. 시간 발전 상태[14] 및 화학에서 영감을 받은 여러 ansätze 등 다양한 전략을 활용합니다. 효율적인 상태 준비에 집중함으로써 QSCI는 화학 해밀토니안에 대한 양자 계산 비용을 절감하는 동시에 높은 충실도를 유지하고, 양자 상태 샘플링 기법의 잡음 견고성을 활용합니다[15]. QSCI는 또한 더 나은 결과를 위해 더 많은 ansätze를 제공하는 적응형 구성 기법도 지원합니다.
화학 문제에 대한 QSCI의 기본 워크플로우는 다음과 같습니다.
- SciPy 등 원하는 소프트웨어를 사용하여 분자 해밀토니안을 구성합니다.
- 적절한 초기 상태와 미리 선택된 파라미터 세트를 가진 화학에서 영감을 받은 ansatz를 선택하여 QSCI 알고리즘을 준비합니다.
- 중요한 기저 상태를 샘플링하고 고전 컴퓨터로 해밀토니안을 대각화하여 바닥 상태 에너지를 구합니다.
- 후처리 기법으로 구성 복구[16]와 대칭 후선택[15]을 사용하는 경우가 많습니다.
- 선택적으로 적응형 QSCI 워크플로우는 무작위 초기 상태와 더 많은 ansätze를 사용하여 2단계에서 3단계까지의 추가 최적화 루프를 포함합니다.
이해도 확인
VQE가 위에 나열된 다른 모든 방법들(상세히 설명되지 않은 QPE 제외)과 공통으로 가지고 있는 점은 무엇인가요?
Answer
모든 방법은 어떤 형태로든 시도 상태 또는 파동함수를 사용합니다. 모든 방법은 이 시도 상태에 대한 초기 추정값이 우수할 때 가장 잘 작동합니다.
또 다른 올바른 답은, 해밀토니안을 측정하기 쉬울 때(비교적 적은 수의 교환 가능한 Pauli 연산자 그룹으로 분류할 수 있을 때) 모두 구현하기 가장 쉽다는 것입니다.
VQE가 위에 나열된 다른 어떤 방법과도 공통점이 없는 점은 무엇인가요?
Answer
고전 최적화 알고리즘입니다. 다른 방법들 중 어느 것도 변분 파라미터를 선택하기 위해 고전 최적화 알고리즘을 사용하지 않습니다.
References
[2] https://en.wikipedia.org/wiki/Variational_quantum_eigensolver
[3] https://journals.aps.org/prapplied/abstract/10.1103/PhysRevApplied.19.024047
[4] https://arxiv.org/abs/2111.05176
[6] https://inquanto.quantinuum.com/tutorials/InQ_tut_fe4n2_2.html
[7] https://www.nature.com/articles/s41467-019-10988-2
[8] https://arxiv.org/abs/2210.15438
[9] https://journals.aps.org/prresearch/abstract/10.1103/PhysRevResearch.6.013254
[10] https://arxiv.org/html/2403.09624v1
[11] https://www.nature.com/articles/s42005-023-01312-y
[13] https://arxiv.org/abs/1802.00171
[14] https://arxiv.org/abs/2103.08505
[15] https://arxiv.org/html/2501.09702v1
[16] https://quri-sdk.qunasys.com/docs/examples/quri-algo-vm/qsci/
[17] https://arxiv.org/abs/2412.13839